2024-07-12
한어Русский языкEnglishFrançaisIndonesianSanskrit日本語DeutschPortuguêsΕλληνικάespañolItalianoSuomalainenLatina
Das GOPlot-Paket dient der Visualisierung biologischer Daten. Vielmehr integriert und visualisiert dieses Paket Expressionsdaten mit den Ergebnissen funktionaler Analysen.Aber sei vorsichtigDieses Paket kann nicht zur Durchführung dieser Analysen, sondern nur zur Visualisierung der Ergebnisse verwendet werden. . In allen Bereichen der Wissenschaft ist es aufgrund von Platzmangel und der für Ergebnisse erforderlichen Einfachheit schwierig, Dinge realistisch zu beschreiben. Daher müssen Informationen visualisiert und Bilder zur Informationsvermittlung verwendet werden. Gut gestaltete Grafiken bieten mehr Informationen auf weniger Platz. Die Idee des Pakets besteht darin, Benutzern die schnelle Untersuchung großer Datenmengen zu ermöglichen, Trends in den Daten aufzudecken und Muster und Korrelationen in den Daten zu finden.
Die Datenvisualisierung kann uns helfen, Antworten auf biologische Fragen zu finden, eine bestimmte Hypothese zu beurteilen und sogar verschiedene Blickwinkel für die Untersuchung verschiedener Probleme zu entdecken. Und die Darstellungsfunktionen dieses Pakets werden auf der Grundlage der hierarchischen Struktur der Daten entwickelt, beginnend mit den Gesamtdaten und endend mit einer Teilmenge ausgewählter Gene und entsprechender Signalwege.
Lassen Sie es uns anhand eines Beispiels konkret erklären.
Wir nennen die mit GOplot gelieferten Daten, die von GEO stammenGSE47067, enthält Transkriptominformationen von Endothelzellen aus zwei Geweben (Gehirn und Herz). Weitere Informationen finden Sie im Artikel von Nolan et al. https://www.ncbi.nlm.nih.gov/pubmed/23871589Die Daten werden normalisiert und es werden unterschiedlich exprimierte Gene gefunden., und verwenden Sie dann das DAVID-Funktionsanmerkungstool (DAVID-Anmerkungsdaten werden langsam aktualisiert und werden derzeit nicht empfohlen. Die Verwendung wird empfohlenGo East, das beste Online-Tool zur GO-AnreicherungsanalyseUndDiese Website, die eine Anreicherungsanalyse in nur einem Schritt durchführen kann, wurde vor ihrer Veröffentlichung mehr als 350 Mal von CNS und anderen zitiert.Anreicherungsanalyse durchführen,Meistern Sie GSEA in einem Artikel, super detailliertes Tutorial) Genannotation von differentiell exprimierten Genen (adjusted p-value < 0.05 ) und funktionelle Anreicherungsanalyse. Dieser Datensatz enthält die folgenden fünf Datenkategorien:
| Name | beschreiben | Datensatzgröße |
|---|---|---|
| EC$eset | Normalisierte Genexpression in Gehirn- und Herzendothelzellen (3 Replikate) | 20644 x 7 |
| EC$Geneliste | Differenziell exprimierte Gene (angepasster p-Wert < 0,05) | 2039 x 7 |
| EC$david | Ergebnisse der funktionellen Anreicherungsanalyse differenzieller Gene mit DAVID | 174 x 5 |
| EC$Gen | Gene und logFC | 37 x 2 |
| EC$Prozess | Ausgewählte Merkmalsvektoren für angereicherte biologische Prozesse | 7 |
Wir möchten die GO-angereicherten Pfade unterschiedlich exprimierter Gene sehen, aber bevor wir mit dem Zeichnen beginnen, müssen wir Daten bereitstellen, die den Formatanforderungen entsprechen.Im Allgemeinen werden die zum Zeichnen des Diagramms erforderlichen Daten von Ihnen selbst bereitgestellt, aberIn diesem Paket gibt es eine Funktioncircle_datKann uns beim Umgang mit dem Datenformat helfen。circle_datEs kann die Ergebnisse der funktionellen Anreicherungsanalyse ausgewählter Gene und ihre logFC-Werte kombinieren, hauptsächlich für unterschiedlich exprimierte Gene.circle_dat Die Verwendung ist sehr einfach, lesen Sie einfach zwei Daten ein. Die ersten Daten enthalten die Ergebnisse der funktionellen Anreicherungsanalyse mit mindestens vier Spalten (Kategorie der funktionellen Anreicherungsanalyse, Signalweg, Gen, angepasster p-Wert).Die zweiten Daten beziehen sich auf das ausgewählte Gen und seinen logFC. Diese Daten können als Quelle dienenlimmaDie Ergebnisse der statistischen Analyse (Anmerkung aus Biografien: Achten Sie unbedingt auf zwei DateienWie Gene benannt werdenSeien Sie konsequent, wie alleGene symbol ). Schauen wir uns die oben genannten Datenformate anhand von Beispielen an.
- #安装已发布的稳定版本
- #install.packages('GOplot')
- #安装github上的开发版本
- #install_github('wencke/wencke.github.io')
- #载入包
- library(GOplot)
- #读入包内自带的数据
- data(EC)
- #查看功能富集分析结果的数据格式
- head(EC$david)
-
- ## Category ID Term
- ## 1 BP GO:0007507 heart development
- ## 2 BP GO:0001944 vasculature development
- ## 3 BP GO:0001568 blood vessel development
- ## 4 BP GO:0048729 tissue morphogenesis
- ## 5 BP GO:0048514 blood vessel morphogenesis
- ## 6 BP GO:0051336 regulation of hydrolase activity
- ## Genes
- ## 1 DLC1, NRP2, NRP1, EDN1, PDLIM3, GJA1, TTN, GJA5, ZIC3, TGFB2, CERKL, GATA6, COL4A3BP, GAB1, SEMA3C, MKL2, SLC22A5, MB, PTPRJ, RXRA, VANGL2, MYH6, TNNT2, HHEX, MURC, MIB1, FOXC2, FOXC1, ADAM19, MYL2, TCAP, EGLN1, SOX9, ITGB1, CHD7, HEXIM1, PKD2, NFATC4, PCSK5, ACTC1, TGFBR2, NF1, HSPG2, SMAD3, TBX1, TNNI3, CSRP3, FOXP1, KCNJ8, PLN, TSC2, ATP6V0A1, TGFBR3, HDAC9
- ## 2 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, EFNB2, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 3 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 4 DLC1, ENAH, NRP1, PGF, ZIC2, TGFB2, CD44, ILK, SEMA3C, RET, AR, RXRA, VANGL2, LEF1, TNNT2, HHEX, MIB1, NCOA3, FOXC2, FOXC1, TGFB1I1, WNT5A, COBL, BBS4, FGFR3, TNC, BMPR2, CTNND1, EGLN1, NR3C1, SOX9, TCF7L1, IGF1R, FOXQ1, MACF1, HOXA5, BCL2, PLXND1, CAR2, ACTC1, TBX4, SMAD3, FZD3, SHANK3, FZD6, HOXB4, FREM2, TSC2, ZIC5, TGFBR3, APAF1
- ## 5 GNA13, CAV1, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, CDH2, MEIS1, WT1, TGFB2, WARS, PTK2, CERKL, APOE, CXCR4, ANG, SEMA3C, PLCD1, NOS2, MKL2, PLXND1, FIGF, FGF2, PTPRJ, TGFBR2, TBX4, NF1, TBX1, TNNI3, PNPLA6, VASH1, THY1, NUS1, MEOX2, ECSCR, AMOT, HBEGF, FOXC2, FOXC1, HDAC7
- ## 6 CAV1, XIAP, AGFG1, ADORA2A, TNNC1, TBC1D9, LEPR, ABHD5, EDN1, ASAP2, ASAP3, SMAP1, TBC1D12, ANG, TBC1D14, MTCH1, TBC1D13, TBC1D4, TBC1D30, DHCR24, HIP1, VAV3, NOS1, NF1, MYH6, RICTOR, TBC1D22A, THY1, PLCE1, RNF7, NDEL1, CHML, IFT57, ACAP2, TSC2, ERN1, APAF1, ARAP3, ARAP2, ARAP1, HTR2A, F2R
- ## adj_pval
- ## 1 0.000002170
- ## 2 0.000010400
- ## 3 0.000007620
- ## 4 0.000119000
- ## 5 0.000720000
- ## 6 0.001171166
-
- #查看基因的数据格式
- head(EC$genelist)
-
- ## ID logFC AveExpr t P.Value adj.P.Val B
- ## 1 Slco1a4 6.645388 1.2168670 88.65515 1.32e-18 2.73e-14 29.02715
- ## 2 Slc19a3 6.281525 1.1600468 69.95094 2.41e-17 2.49e-13 27.62917
- ## 3 Ddc 4.483338 0.8365231 65.57836 5.31e-17 3.65e-13 27.18476
- ## 4 Slco1c1 6.469384 1.3558865 59.87613 1.62e-16 8.34e-13 26.51242
- ## 5 Sema3c 5.515630 2.3252117 58.53141 2.14e-16 8.81e-13 26.33626
- ## 6 Slc38a3 4.761755 0.9218670 54.11559 5.58e-16 1.76e-12 25.70308
Nachdem Sie die beiden Eingabedatenformate verstanden haben, können Sie sie verwendencirlce_datFunktion zum Generieren von Zeichnungsdaten.
- # 生成画图所需的数据格式
- circ <- circle_dat(EC$david, EC$genelist)
-
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
circDas Objekt verfügt über acht Datenspalten, nämlich
Kategorie: BP (Biological Process), CC (Cellular Component) oder MF (Molecular Function)
ID: GO-ID (optionale Spalte, wenn Sie ein Funktionsanalysetool verwenden möchten, das nicht auf der GO-ID basiert, können Sie die ID-Spalte nicht auswählen; die ID kann hier auch KEGG-ID sein)
Begriff: GO-Pfad
count: Anzahl der Gene in jedem Signalweg
gene: Genname - logFC: logFC-Wert jedes Gens
adj_pval: angepasster p-Wert, Pfade mit adj_pval<0,05 gelten als deutlich angereichert
zscore: zscore bezieht sich nicht auf eine statistische Normalisierungsmethode, sondern ist ein leicht zu berechnender Wert, um abzuschätzen, ob ein biologischer Prozess (/molekulare Funktion/zelluläre Komponente) eher abnimmt (negativer Wert) oder zunimmt (positiver Wert).Die Berechnungsmethode ist die Anzahl der hochregulierten Gene minus der Anzahl der herunterregulierten Gene dividiert durch die Quadratwurzel der Anzahl der Gene in jedem Signalweg.
Wenn wir uns die Daten zum ersten Mal ansehen, möchten wir so viele Pfade wie möglich aus dem Diagramm anzeigen und auch wertvolle Pfade finden. Daher benötigen wir einige Parameter, um die Wichtigkeit zu bewerten. Balkendiagramme werden häufig zur Beschreibung von Beispieldaten verwendet, sodass wir mit der GOBar-Funktion schnell ein ansprechendes Balkendiagramm erstellen können.
Zunächst wird direkt ein einfaches Balkendiagramm generiertGO Terms, nach IhnenzscoreSortieren Sie die Balken; die vertikale Achse ist-log(adj p-value);Die Farbe repräsentiertzscore, blau zeigt anz-scoreist ein negativer Wert, ist es wahrscheinlicher, dass die Genexpression im entsprechenden Signalweg abnimmt, was in Rot angezeigt wirdz-score ist ein positiver Wert, ist es wahrscheinlicher, dass die Genexpression im entsprechenden Signalweg zunimmt. Bei Bedarf kann die Reihenfolge geändert werden, indem der Parameter order.by.zscore auf FALSE gesetzt wird. In diesem Fall werden die Balken nach ihrer Bedeutung sortiert.
- # 生成简单的条形图
- GOBar(subset(circ, category == 'BP'))
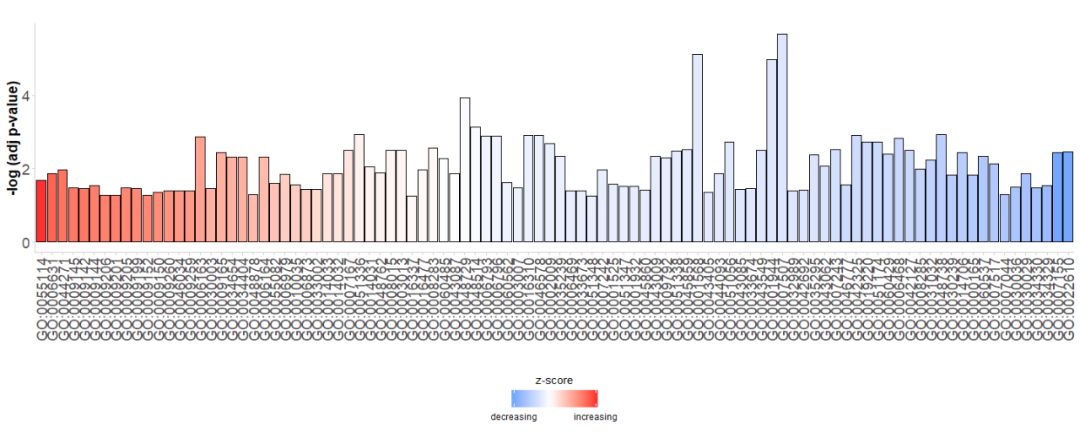
#GOBar(subset(circ, category == 'BP',order.by.zscore=FALSE))Ändern Sie außerdem den Anzeigeparameter, um ein Balkendiagramm entsprechend der Kategorie des Kanals zu zeichnen.
- #根据通路的类别来绘制条形图
- GOBar(circ, display = 'multiple')
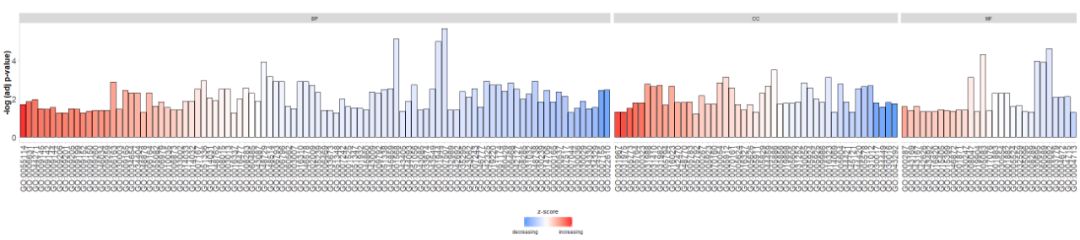
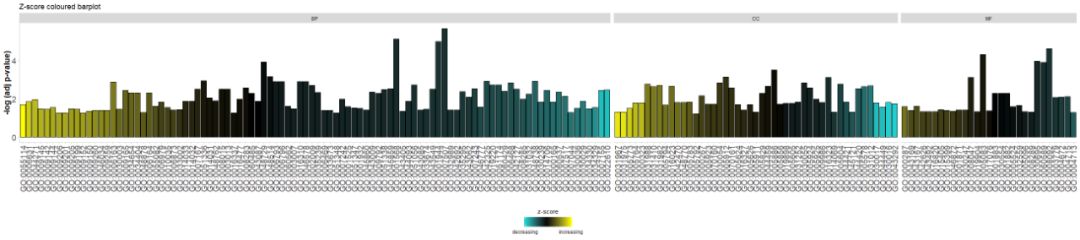
Fügen Sie einen Titel hinzu und verwenden Sie Parameterzsc.colÄndernzscores Farbe.
- # Facet the barplot, add a title and change the colour scale for the z-score
- GOBar(circ, display = 'multiple', title = 'Z-score coloured barplot', zsc.col = c('yellow', 'black', 'cyan'))
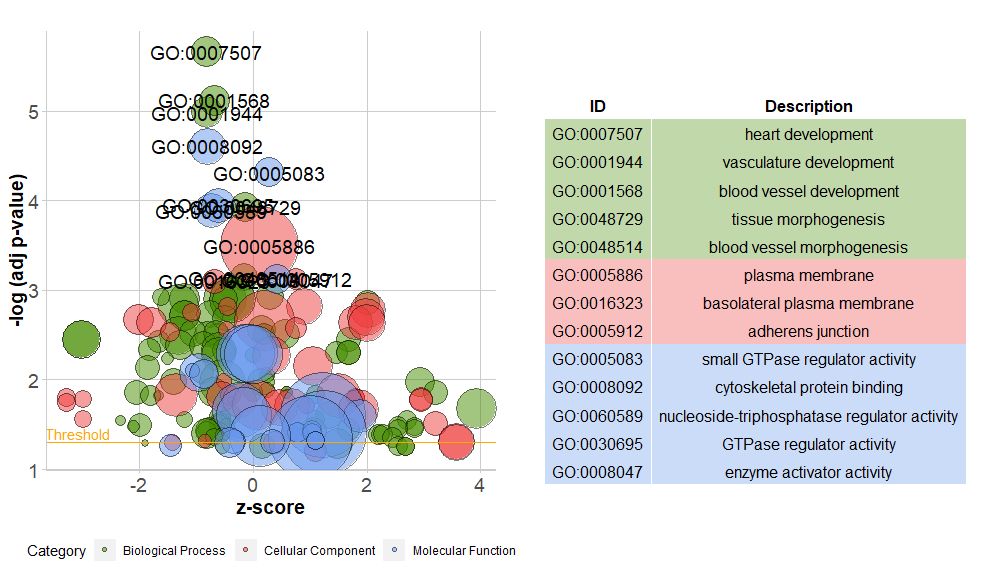
Balkendiagramme sind sehr verbreitet und leicht zu verstehen, aber wir können Blasendiagramme verwenden, um mehr Informationen über die Daten anzuzeigen.
Die horizontale Achse istzscore;Die vertikale Achse ist-log(adj p-value)Ähnlich wie bei einem Balkendiagramm ist die Anreicherung umso signifikanter, je höher sie ist. Die Fläche des Kreises hängt von der Anzahl der Gene im entsprechenden Pfad ab (circ$count ); die Farbe entspricht der Kategorie, die dem Signalweg entspricht, Grün ist der biologische Prozess, Rot ist die zelluläre Komponente und Blau ist die molekulare Funktion.Kann eingegeben werden von?GOBubble Informationen zum Ändern aller Bildparameter finden Sie auf der Hilfeseite der GOBubble-Funktion. Standardmäßig ist jeder Kreis mit einer entsprechenden GO-ID gekennzeichnet, und rechts wird außerdem eine Tabelle angezeigt, die die entsprechende Beziehung zwischen GO-ID und GO-Begriff zeigt.Parameter können eingestellt werden durchtable.legendfürFALSE um es zu verbergen. Wenn Sie die Pfadbeschreibung anzeigen möchten, setzen Sie den Parameter ID auf FALSE.Aufgrund des begrenzten Platzes und der überlappenden Kreise werden jedoch nicht alle Kreise markiert, sondern nur die-log(adj p-value) > 3(Standard ist 5).
- # 生成泡泡图,并展示-log(adj p-value) > 3 的通路的GO ID
- GOBubble(circ, labels = 3)
Wenn Sie dem Blasendiagramm einen Titel hinzufügen oder die Farbe des Kreises angeben und die Pfade jeder Kategorie separat anzeigen und den angezeigten GO-ID-Schwellenwert ändern möchten, können Sie die folgenden Parameter hinzufügen:
GOBubble(circ, title = 'Bubble plot', colour = c('orange', 'darkred', 'gold'), display = 'multiple', labels = 3)
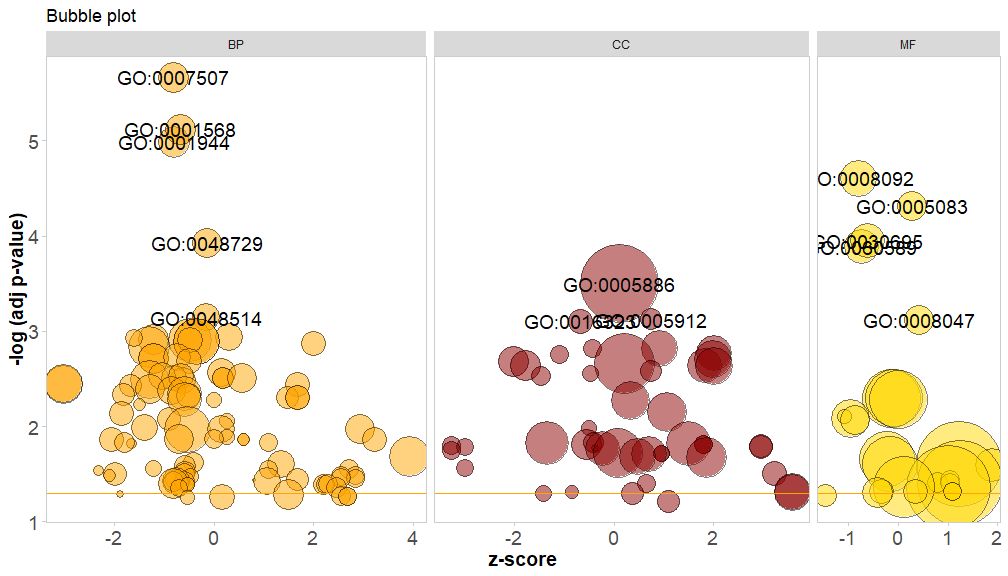
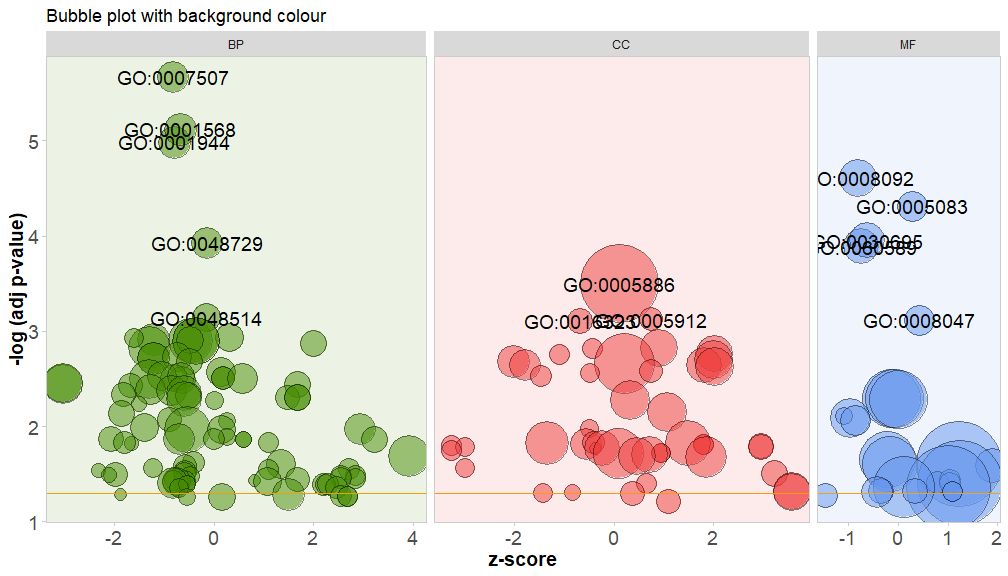
Färben Sie den Hintergrund der Kanalklasse, indem Sie den Parameter bg.col auf TRUE setzen.
GOBubble(circ, title = 'Bubble plot with background colour', display = 'multiple', bg.col = T, labels = 3)
Die neue Version des Pakets enthält eine neue Funktionreduce_overlap Diese Funktion kann die Anzahl redundanter Elemente reduzieren, d. h. sie kann alle Pfade löschen, deren Genüberlappung größer oder gleich dem festgelegten Schwellenwert ist, und nur einen Pfad aus jeder Gruppe als Vertreter beibehalten, unabhängig von der Anzeige aller Pfade in GO. Durch die Reduzierung der Anzahl redundanter Terme wird die Lesbarkeit von Diagrammen (z. B. Blasendiagrammen) erheblich verbessert.
- # reduce_overlap,参数设置为0.75
- reduced_circ <- reduce_overlap(circ, overlap = 0.75)
-
- GOBubble(reduced_circ, labels = 2.8)
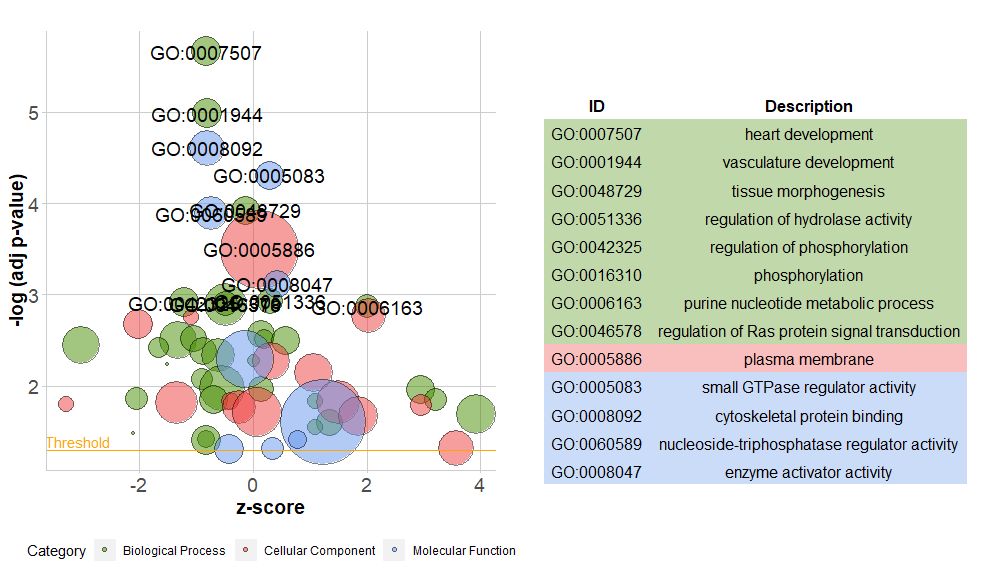
Obwohl uns eine Grafik mit allen Informationen dabei helfen kann, herauszufinden, welche Pfade am sinnvollsten sind, hängt die Realität immer noch von den Hypothesen und Ideen ab, die Sie mit den Daten bestätigen möchten, und die wichtigsten Pfade sind möglicherweise nicht unbedingt diejenigen, an denen Sie interessiert sind. Daher ist bei der manuellen Auswahl eines wertvollen Satzes von Pfaden (EC$process ), benötigen wir ein Diagramm, das uns detailliertere Informationen über diese spezifische Gruppe von Pfaden zeigt.Bei der Darstellung dieser Zahlen entsteht jedoch ein Problem: Manchmal ist die Interpretation schwierigzscore Informationen bereitgestellt.Schließlich ist diese Berechnungsmethode nicht universell. Wie oben gezeigt, wird einfach die Anzahl der hochregulierten Gene minus der Anzahl der herunterregulierten Gene durch die Quadratwurzel der Anzahl der Gene in jedem Pfad dividiertGOCircleAuch die resultierende Grafik unterstreicht diese Tatsache.
Der äußere Kreis des Kreisdiagramms zeigt den logFC-Wert der Gene jedes Signalwegs als verstreute Punkte. Rote Kreise zeigen eine Hochregulierung und blaue Kreise eine Herunterregulierung an.Parameter können verwendet werdenlfc.col Farbe ändern. Dies erklärt auch, warum in einigen Fällen sehr wichtige Pfade Z-Scores nahe Null aufweisen. Ein Zscore von Null bedeutet nicht, dass der Kanal unwichtig ist. Es zeigt lediglich, dass es sich bei Zscore um ein grobes Maß handelt, da Zscore offensichtlich auch nicht das Funktionsniveau und die Aktivierungsabhängigkeit einzelner Gene in biologischen Prozessen berücksichtigt.
GOCircle(circ)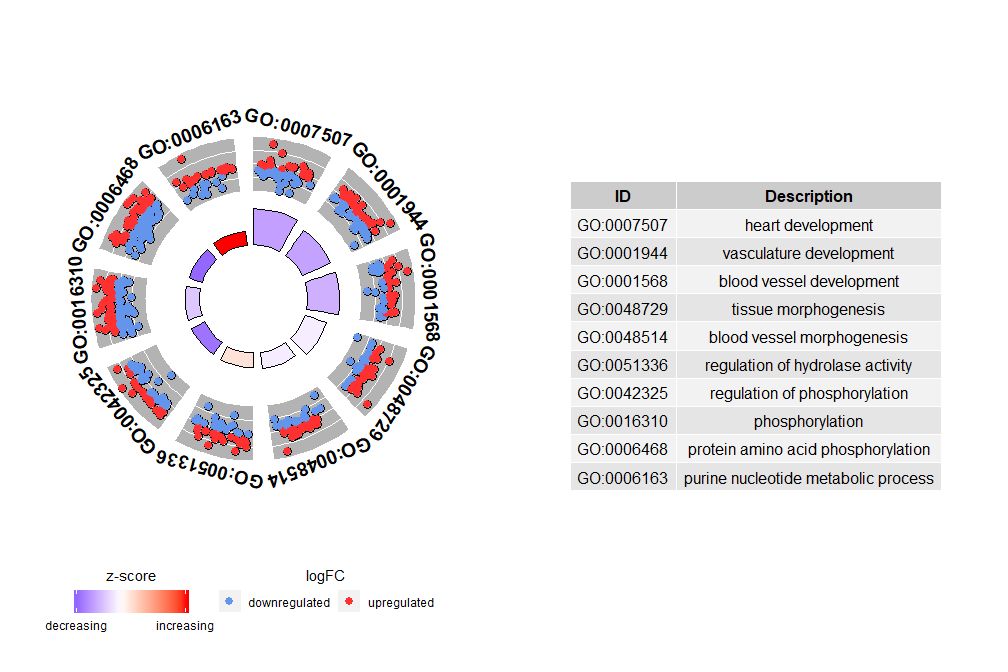
nsub Parameter können eingestellte Zahlen oder Zeichenvektoren sein. Wenn es sich um einen Zeichenvektor handelt, enthält er die GO-ID oder den anzuzeigenden Pfad.
- # 生成特定通路的圈图
- IDs <- c('GO:0007507', 'GO:0001568', 'GO:0001944', 'GO:0048729', 'GO:0048514', 'GO:0005886', 'GO:0008092', 'GO:0008047')
- GOCircle(circ, nsub = IDs)
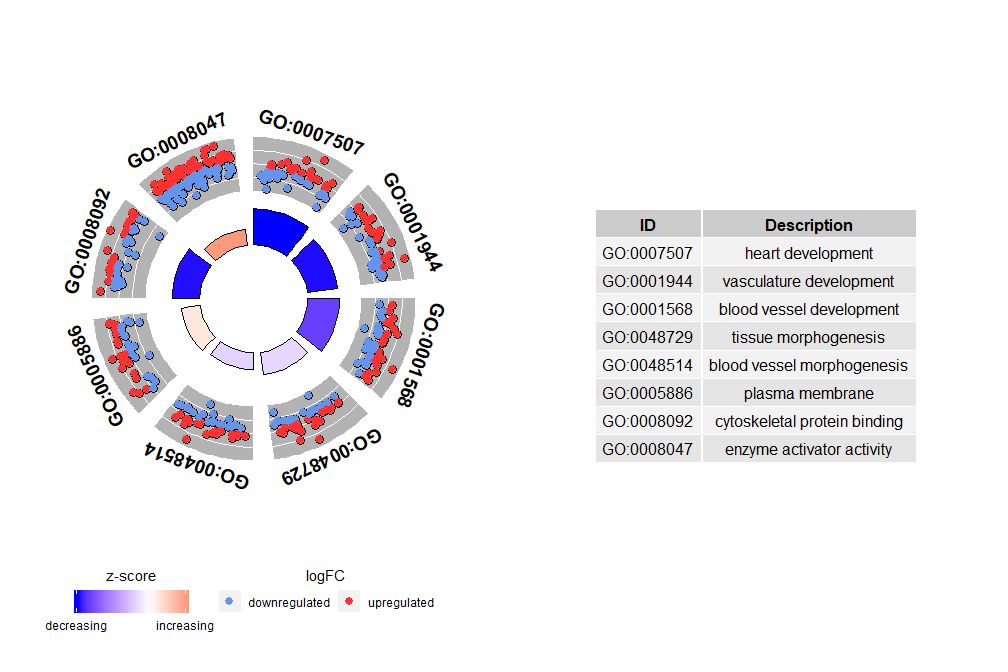
Wenn nsub ein numerischer Vektor ist, definiert die Zahl die anzuzeigende Zahl. Es beginnt in der ersten Zeile des Eingabedatenrahmens. Diese Visualisierung funktioniert nur mit kleineren Daten. Die maximale Anzahl der Kanäle beträgt standardmäßig 12. Obwohl die Anzahl der Kanäle reduziert wird, erhöht sich die Menge der angezeigten Informationen.
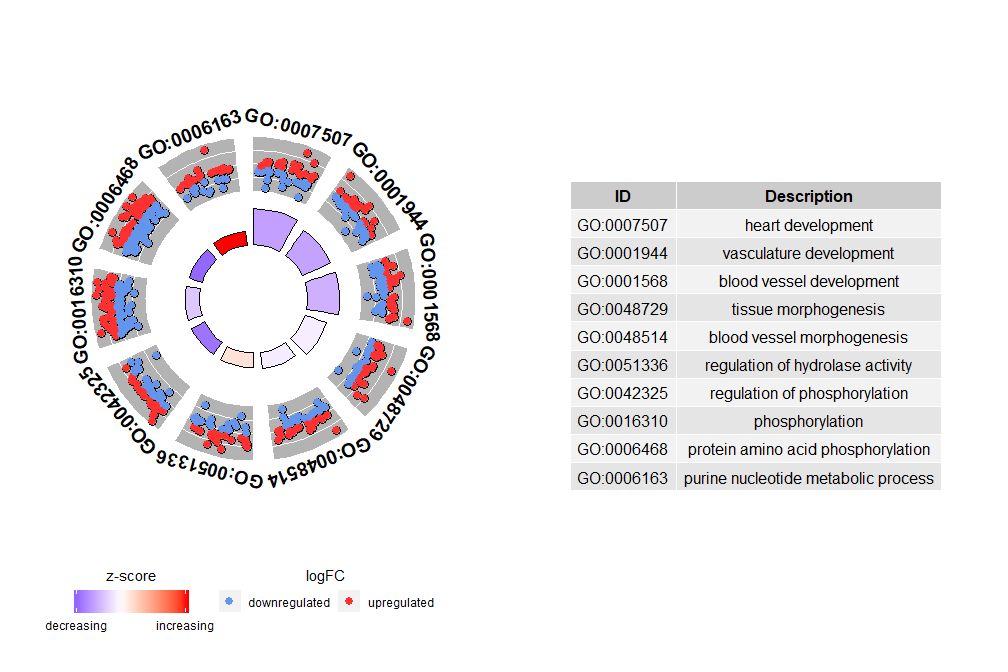
- # 圈图展示数据前十个通路
- GOCircle(circ, nsub = 10)
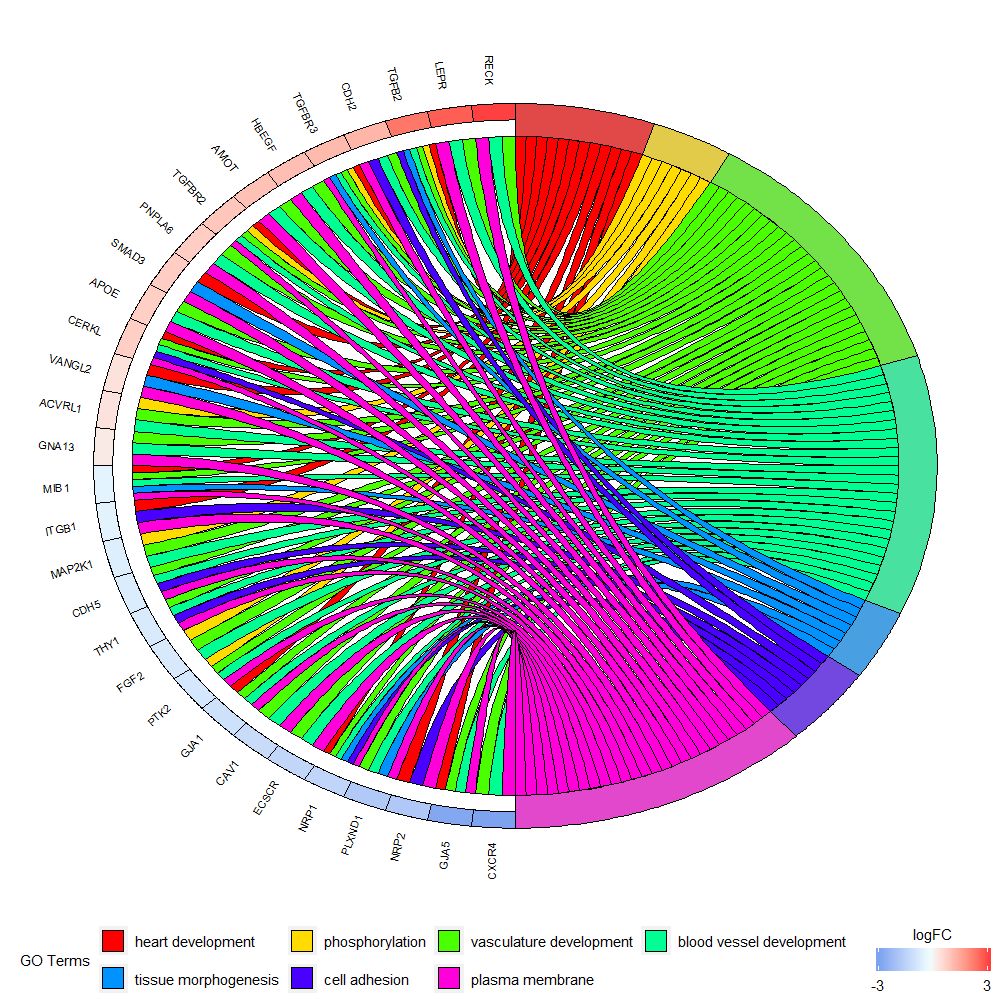
GOChord kann die Beziehung zwischen ausgewählten Genen und Signalwegen sowie den logFC der Gene anzeigen.Zuerst müssen Sie eine Matrix eingeben, die Sie selbst erstellen können0-1Matrix können Sie auch Funktionen verwendenchord_dat Bauen. Diese Funktion verfügt über drei Parameter: Daten, Gene und Prozess, wobei die letzten beiden Parameter mindestens einen Parameter haben müssen.Dann die Funktioncircle_datKombinieren Sie Expressionsdaten mit Ergebnissen aus Funktionsanalysen.
Balkendiagramme und Blasendiagramme können Ihnen einen ersten Eindruck von den Daten vermitteln. Jetzt können Sie einige Gene und Pfade auswählen, die unserer Meinung nach wertvoll sind. Obwohl GOCircle eine Ebene hinzufügt, um den Expressionswert von Genen in Pfaden anzuzeigen, fehlen einzelne Informationen dazu Beziehungen zwischen Genen und mehreren Signalwegen. Es ist nicht einfach herauszufinden, ob bestimmte Gene mit mehreren Prozessen verknüpft sind. GOChord gleicht die Mängel von GOCircle aus. Die Zeilen der generierten Daten sind Gene und die Spalten sind Pfade. „0“ bedeutet, dass das Gen dem Pfad nicht zugeordnet ist, und „1“ ist das Gegenteil.
- # 找到感兴趣的的基因,这里我们以EC$genes为例
- head(EC$genes)
-
- ## ID logFC
- ## 1 PTK2 -0.6527904
- ## 2 GNA13 0.3711599
- ## 3 LEPR 2.6539788
- ## 4 APOE 0.8698346
- ## 5 CXCR4 -2.5647537
- ## 6 RECK 3.6926860
-
- # 获得感兴趣基因的通路
- EC$process
-
- ## [1] "heart development" "phosphorylation"
- ## [3] "vasculature development" "blood vessel development"
- ## [5] "tissue morphogenesis" "cell adhesion"
- ## [7] "plasma membrane"
-
- # 使用chord_dat构建矩阵
- chord <- chord_dat(circ, EC$genes, EC$process)
- head(chord)
-
- ## heart development phosphorylation vasculature development
- ## PTK2 0 1 1
- ## GNA13 0 0 1
- ## LEPR 0 0 1
- ## APOE 0 0 1
- ## CXCR4 0 0 1
- ## RECK 0 0 1
- ## blood vessel development tissue morphogenesis cell adhesion
- ## PTK2 1 0 0
- ## GNA13 1 0 0
- ## LEPR 1 0 0
- ## APOE 1 0 0
- ## CXCR4 1 0 0
- ## RECK 1 0 0
- ## plasma membrane logFC
- ## PTK2 1 -0.6527904
- ## GNA13 1 0.3711599
- ## LEPR 1 2.6539788
- ## APOE 1 0.8698346
- ## CXCR4 1 -2.5647537
- ## RECK 1 3.6926860
Im Beispiel haben wir zwei Parameter übergeben. Wenn nur der Genes-Parameter angegeben wird, ist das Ergebnis eine Liste ausgewählter Gene und aller Prozesskonstruktionen mit mindestens einem angegebenen Gen.0-1Matrix; wenn nur angegebenprocessParameter, das Ergebnis ist, dass alle Gene generiert werden0-1 Matrix von Genen, die mindestens einem Prozess in der Liste zugeordnet sind. Beachten Sie, dass die Angabe nur der Gene und Prozessparameter zu einer sehr großen 0-1-Matrix führen kann, was zu verwirrenden Visualisierungsergebnissen führt.
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
-
- # Generate the matrix with a list of selected genes
- chord_genes <- chord_dat(data = circ, genes = EC$genes)
- head(chord_genes)
-
- ## heart development vasculature development blood vessel development
- ## PTK2 0 1 1
- ## GNA13 0 1 1
- ## LEPR 0 1 1
- ## APOE 0 1 1
- ## CXCR4 0 1 1
Dieses Diagramm soll eine kleinere Teilmenge hochdimensionaler Daten zeigen. Es gibt hauptsächlich zwei Parameter, die angepasst werden können:gene.orderUndnlfc . Der Genes-Parameter kann als „logFC“, „alphabetisch“, „none“ angegeben werden. Tatsächlich geben wir den Genes-Parameter im Allgemeinen als logFC an. Der nlfc-Parameter ist einer der wichtigsten Parameter dieser Funktion, da er verarbeiten kann, wie jedes Gen 0 oder mehr logFC-Werte in der Matrix aufweist. Daher sollten wir Parameter angeben, um Fehler zu vermeiden.
Wenn Sie beispielsweise eine Matrix ohne logFC-Werte haben, müssen Sie festlegennlfc=0 ; Oder führen Sie eine differenzielle Expressionsanalyse für Gene unter mehreren Bedingungen oder Chargen durch. In diesem Fall enthält jedes Gen mehrere logFC-Werte und die Spaltennummer nlfc=logFC muss festgelegt werden. Der Standardwert ist „1“, da davon ausgegangen wird, dass es in den meisten Fällen nur einen logFC-Wert pro Gen gibt. Verwenden Sie den Parameter „space“, um den Abstand zwischen farbigen Rechtecken zu definieren, die logFC darstellen. Der Parameter gene.size gibt die Schriftgröße des Gennamens an, und gene.space gibt die Leerraumgröße zwischen den Gennamen an.
- chord <- chord_dat(data = circ, genes = EC$genes, process = EC$process)
- GOChord(chord, space = 0.02, gene.order = 'logFC', gene.space = 0.25, gene.size = 5)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
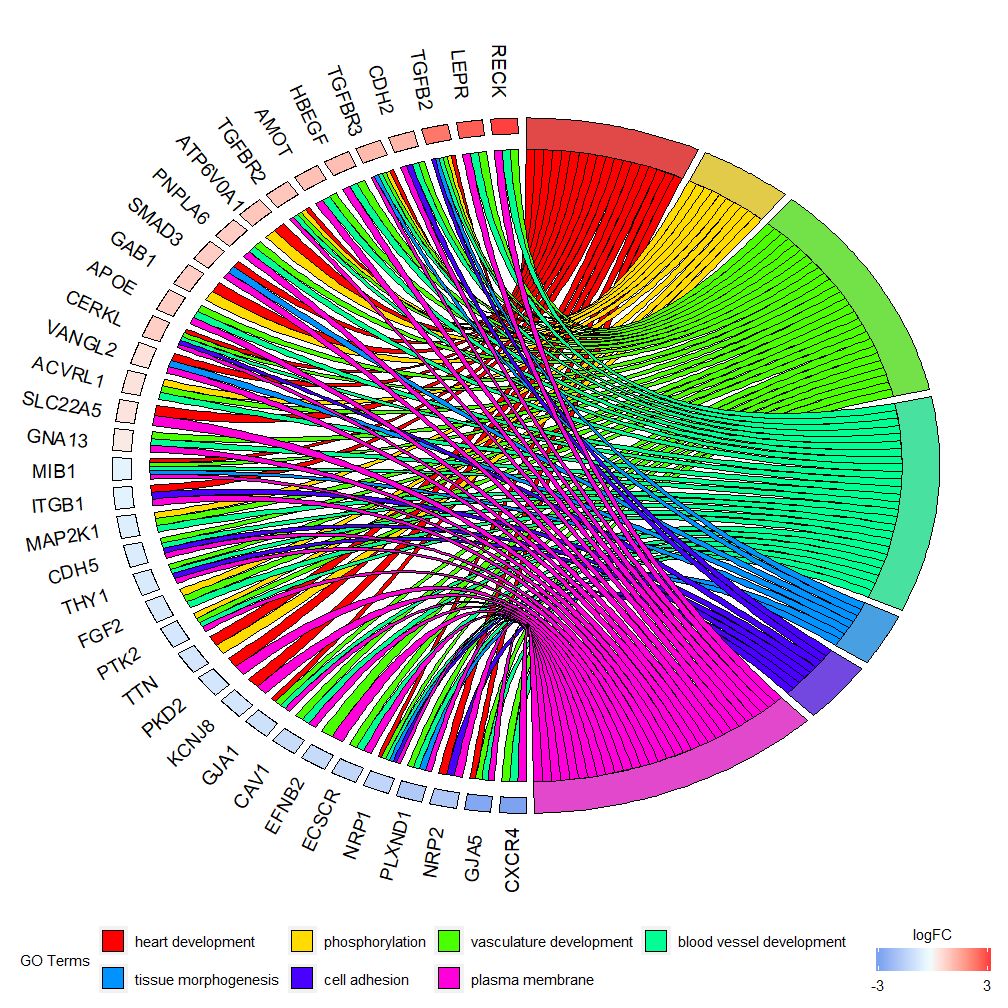
Kann entsprechend dem logFC-Wert eingestellt werdengene.order=‘logFC’ Sortieren Sie Gene nach ihren logFC-Werten. Manchmal kann das Bild etwas unübersichtlich werden. Dies kann automatisiert werden, indem der Parameter „Limit“ verwendet wird, um die Anzahl der angezeigten Gene oder Pfade zu reduzieren. Limit ist ein Vektor mit zwei Grenzwerten (der Standardwert ist c(0,0)). Der erste Wert gibt die Mindestanzahl an Signalwegen an, denen das Gen zugeordnet werden muss. Der zweite Wert bestimmt die Anzahl der dem Signalweg zugeordneten Gene.
- # 仅显示分配给至少三个通路的基因
- GOChord(chord, limit = c(3, 0), gene.order = 'logFC')
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
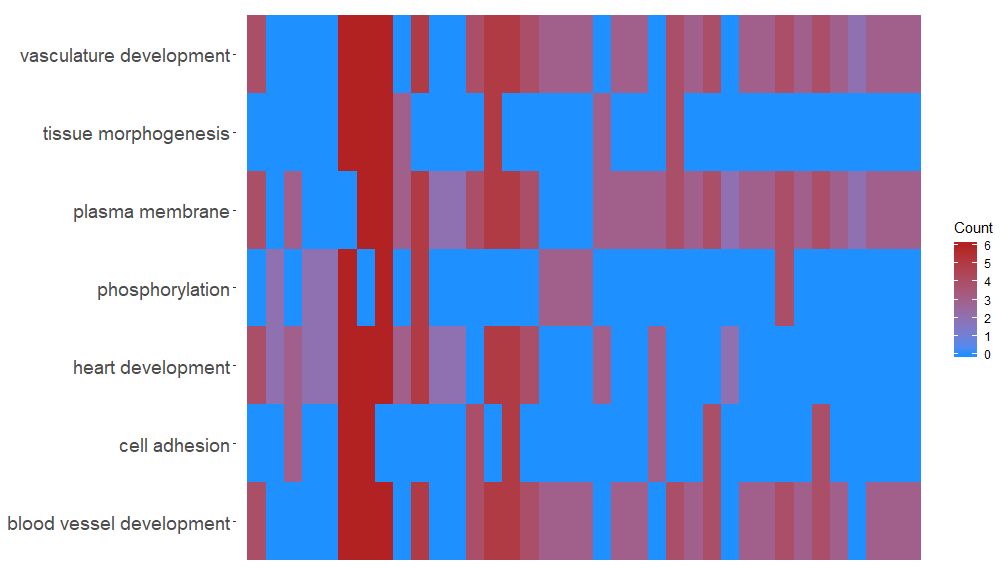
Die GOHeat-Funktion kann die Beziehung zwischen Genen und Signalwegen mithilfe einer Heatmap anzeigen, ähnlich wie GOChord. Biologische Prozesse werden horizontal und Gene vertikal dargestellt. Jede Spalte ist in kleine Rechtecke unterteilt und die Farbe hängt im Allgemeinen vom logFC-Wert ab. Darüber hinaus wurden Gene geclustert, die an ähnlichen Funktionswegen angereichert sind. Abhängig von den NFC-Parametern gibt es zwei Modi für die Farbauswahl der Heatmap. Wenn nlfc = 0, ist die Farbe die Anzahl der angereicherten Pfade für jedes Gen. Einzelheiten finden Sie in den Beispielen:
- # First, we use the chord object without logFC column to create the heatmap
- GOHeat(chord[,-8], nlfc = 0)
GOHeat(chord[,-8])Die Farbe entspricht logFC des Gens im Fall nlfc = 1
GOHeat(chord, nlfc = 1, fill.col = c('red', 'yellow', 'green'))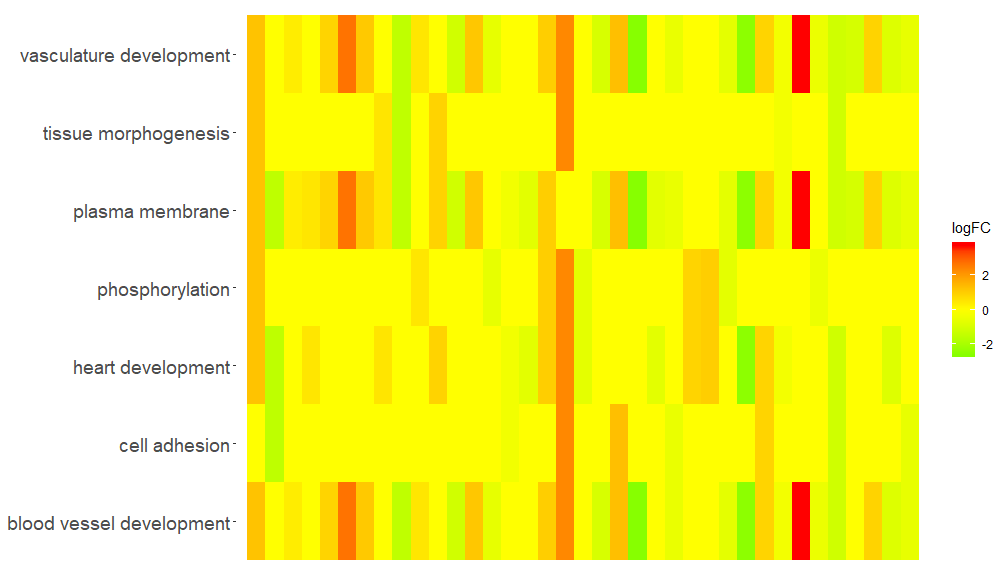
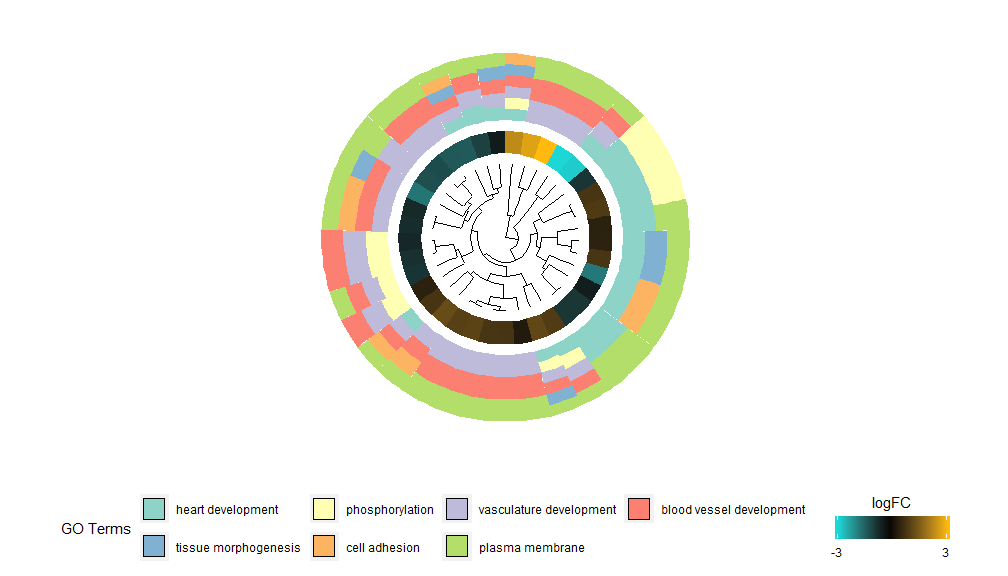
Die Idee hinter der GOCluster-Funktionalität besteht darin, möglichst viele Informationen anzuzeigen. Hier ist ein Beispiel:
- GOCluster(circ, EC$process, clust.by = 'logFC', term.width = 2)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
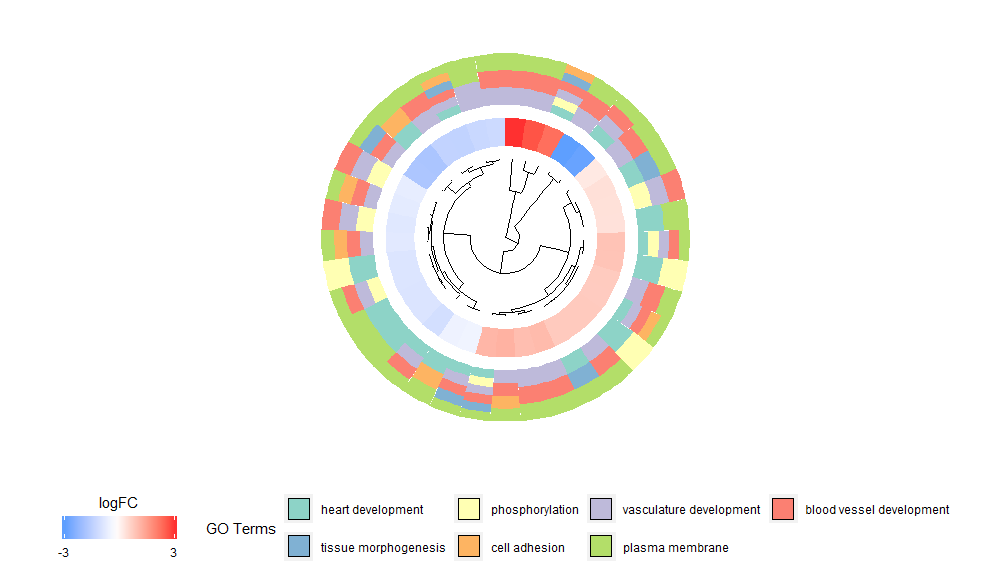
Hierarchisches Clustering ist eine beliebte unbeaufsichtigte Clusteranalysemethode für die Genexpression, die eine unvoreingenommene Gruppierung von Genen nach Expressionsmuster gewährleistet, sodass Cluster, die zusammen gruppiert werden, mehrere Gruppen koregulierter oder funktionell verwandter Gene enthalten können. GOCluster verwendet diehclust Die Methode führt eine hierarchische Gruppierung von Genexpressionsprofilen durch. Wenn Sie die Distanzmetrik oder den Clustering-Algorithmus ändern möchten, verwenden Sie die Parameter metric bzw. clust. Das resultierende Dendrogramm kann mit Hilfe von ggdendro konvertiert und mit ggplot2 visualisiert werden. Wählen Sie einen kreisförmigen Grundriss, da dieser nicht nur effektiv, sondern auch optisch ansprechend ist. Der erste Kreis neben dem Dendrogramm stellt den logFC des Gens dar, der eigentlich das Blatt des Clustering-Baums ist. Wenn Sie an mehreren Kontrasten interessiert sind, können Sie den Parameter nlfc ändern. Standardmäßig ist er auf „1“ gesetzt, sodass nur ein Ring gezeichnet wird. Die logFC-Werte werden mithilfe einer vom Benutzer definierbaren Farbskala (lfc.col) farblich gekennzeichnet. Der nächste Kreis stellt den dem Gen zugeordneten Pfad dar. Um gut auszusehen, wurde die Anzahl der Kanäle reduziert und die Farbe der Kanäle kann über den Parameter term.col geändert werden.noch verfügbar?GOCluster um zu sehen, wie man die Parameter ändert. Der wichtigste Parameter dieser Funktion ist „cluster.by“, der nach Genexpressionsmustern („logFC“, wie oben gezeigt) oder Funktionskategorien („Begriffe“) gruppiert werden kann.
- GOCluster(circ, EC$process, clust.by = 'term', lfc.col = c('darkgoldenrod1', 'black', 'cyan1'))
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
Venn-Diagramme können verwendet werden, um Beziehungen zwischen verschiedenen Listen unterschiedlich exprimierter Gene zu erkennen oder um die Schnittmenge mehrerer Pathway-Gene in Funktionsanalysen zu untersuchen. Venn-Diagramme zeigen nicht nur die Anzahl überlappender Gene, sondern auch Informationen über das Expressionsmuster des Gens (normalerweise hochreguliert, oft herunterreguliert oder gegenreguliert). Derzeit werden bis zu drei Datensätze als Eingabe verwendet. Die Eingabedaten enthalten mindestens zwei Spalten: eine für Gennamen und eine für logFC-Werte.
- l1 <- subset(circ, term == 'heart development', c(genes,logFC))
- l2 <- subset(circ, term == 'plasma membrane', c(genes,logFC))
- l3 <- subset(circ, term == 'tissue morphogenesis', c(genes,logFC))
- GOVenn(l1,l2,l3, label = c('heart development', 'plasma membrane', 'tissue morphogenesis'))
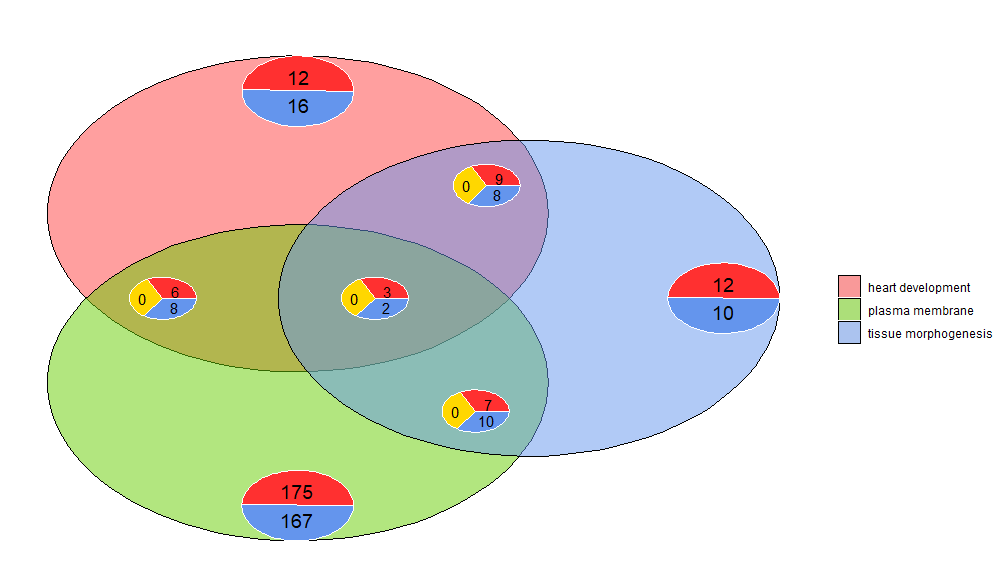
Beispielsweise gibt es für die Herzentwicklung und die Gewebemorphogenese 22 Gene, von denen 12 hochreguliert und 10 herunterreguliert sind. Es ist wichtig zu beachten, dass Kreisdiagramme keine redundanten Informationen anzeigen. Wenn daher drei Datensätze verglichen werden, sind Gene, die allen Datensätzen gemeinsam sind (das mittlere Kreisdiagramm), nicht in den anderen Kreisdiagrammen enthalten. Dieses Tool ist auf Shinyapp https://wwalter.shinyapps.io/Venn/ verfügbar. Das Webtool ist interaktiver, der Kreis hat eine Fläche proportional zur Anzahl der Gene im Datensatz und der Schieberegler kann zum Verschieben verwendet werden kleines Kreisdiagramm und verfügt über alle Optionen von GOVenn Features, um das Layout des Plots zu ändern und auch Bilder und Genlisten herunterzuladen.
Software-Homepage: https://wencke.github.io/

Er widmet sich seit mehr als 30 Jahren der Technologieforschung und beherrscht verschiedene Sprachen wie Java, Linux, Javascript, PHP, CSS usw. Er hat viele Beiträge im Open-Source-Bereich geleistet Entwicklerdokumentationsstation, um einige Themen in der Technologieentwicklung als zukünftige Referenz zu teilen

