2024-07-12
한어Русский языкEnglishFrançaisIndonesianSanskrit日本語DeutschPortuguêsΕλληνικάespañolItalianoSuomalainenLatina
GOPlot-pakettia käytetään biologisten tietojen visualisointiin. Pikemminkin tämä paketti integroi ja visualisoi ilmaisutiedot toiminnallisten analyysien tuloksiin.Mutta ole varovainenTätä pakettia ei voida käyttää näiden analyysien suorittamiseen, vaan tulosten visualisointiin. . Kaikilla tieteenaloilla asioita on vaikea kuvata realistisesti tilanrajoitusten ja tulosten vaatiman yksinkertaisuuden vuoksi, joten tietoa on visualisoitava ja kuvia käytettävä tiedon välittämiseen. Hyvin suunniteltu grafiikka tarjoaa enemmän tietoa pienemmässä tilassa. Paketin ideana on antaa käyttäjille mahdollisuus tutkia nopeasti suuria tietomääriä, paljastaa datan trendejä ja löytää tiedosta malleja ja korrelaatioita.
Datan visualisointi voi auttaa meitä löytämään vastauksia biologisiin kysymyksiin, arvioimaan tiettyä hypoteesia ja jopa löytämään erilaisia näkökulmia erilaisten ongelmien tutkimiseen. Ja tämän paketin piirtofunktiot on kehitetty datan hierarkkisen rakenteen perusteella, alkaen kokonaistiedoista ja päättyen valittujen geenien ja vastaavien reittien osajoukkoon.
Selitetään se konkreettisesti esimerkin avulla.
Kutsumme dataa, joka tulee GOplotista, joka tulee GEO:ltaGSE47067, joka sisältää kahden kudoksen (aivot ja sydän) transkriptiotiedot, katso Nolan et al.Tiedot normalisoidaan ja eri tavalla ilmentyviä geenejä löydetään., ja käytä sitten DAVID-funktion merkintätyökalua (DAVID-merkintätiedot päivittyvät hitaasti, eikä niitä suositella nyt. On suositeltavaa käyttääGo East, paras online-GO-rikastusanalyysityökalujaCNS ja muut ovat lainanneet tätä verkkosivustoa, joka voi suorittaa rikastusanalyysin vain yhdessä vaiheessa, yli 350 kertaa ennen sen julkaisua.Suorita rikastusanalyysi,Hallitse GSEA yhdessä artikkelissa, erittäin yksityiskohtainen opetusohjelma) Erilaisesti ilmentyneiden geenien geenimerkintä (adjusted p-value < 0.05 ) ja toiminnallinen rikastusanalyysi. Tämä tietojoukko sisältää seuraavat viisi tietoluokkaa:
| nimi | kuvata | Tietojoukon koko |
|---|---|---|
| EC$eset | Normalisoitunut geeniekspressio aivojen ja sydämen endoteelisoluissa (3 toistoa) | 20644 x 7 |
| EC$ genelist | Erilaisesti ilmentyvät geenit (säädetty p-arvo < 0,05) | 2039 x 7 |
| EC$ David | Tulokset differentiaalisten geenien toiminnallisesta rikastusanalyysistä DAVIDia käyttämällä | 174 x 5 |
| EC$-geeni | Geenit ja logFC | 37x2 |
| EC$-prosessi | Valitut piirrevektorit rikastettuihin biologisiin prosesseihin | 7 |
Haluamme nähdä differentiaalisesti ilmentyneiden geenien GO-rikastetut reitit, mutta ennen kuin aloitamme piirtämisen, meidän on toimitettava muotovaatimukset täyttävät tiedot.Yleisesti ottaen kaavion piirtämiseen tarvittavat tiedot annat itse, muttaTässä paketissa on toimintocircle_datVoi auttaa meitä käsittelemään tietomuotoa。circle_datSe voi yhdistää valittujen geenien funktionaalisen rikastusanalyysin tulokset ja niiden logFC-arvot, pääasiassa differentiaalisesti ilmentyneiden geenien osalta.circle_dat Käyttö on hyvin yksinkertaista, lue vain kahdessa tiedossa. Ensimmäiset tiedot sisältävät funktionaalisen rikastusanalyysin tulokset vähintään neljällä sarakkeella (funktionaalisen rikastamisen analyysiluokka, reitti, geeni, säädetty p-arvo).Toinen data on valitusta geenistä ja sen logFC:stä, tämä data voi olla lähdelimmaTilastollisen analyysin tulokset (Huomautus Biographiesista: Muista kiinnittää huomiota kahteen tiedostoonKuinka geenit nimetäänOle johdonmukainen, kuten kaikkiGene symbol ). Katsotaanpa edellä mainittuja tietomuotoja esimerkein.
- #安装已发布的稳定版本
- #install.packages('GOplot')
- #安装github上的开发版本
- #install_github('wencke/wencke.github.io')
- #载入包
- library(GOplot)
- #读入包内自带的数据
- data(EC)
- #查看功能富集分析结果的数据格式
- head(EC$david)
-
- ## Category ID Term
- ## 1 BP GO:0007507 heart development
- ## 2 BP GO:0001944 vasculature development
- ## 3 BP GO:0001568 blood vessel development
- ## 4 BP GO:0048729 tissue morphogenesis
- ## 5 BP GO:0048514 blood vessel morphogenesis
- ## 6 BP GO:0051336 regulation of hydrolase activity
- ## Genes
- ## 1 DLC1, NRP2, NRP1, EDN1, PDLIM3, GJA1, TTN, GJA5, ZIC3, TGFB2, CERKL, GATA6, COL4A3BP, GAB1, SEMA3C, MKL2, SLC22A5, MB, PTPRJ, RXRA, VANGL2, MYH6, TNNT2, HHEX, MURC, MIB1, FOXC2, FOXC1, ADAM19, MYL2, TCAP, EGLN1, SOX9, ITGB1, CHD7, HEXIM1, PKD2, NFATC4, PCSK5, ACTC1, TGFBR2, NF1, HSPG2, SMAD3, TBX1, TNNI3, CSRP3, FOXP1, KCNJ8, PLN, TSC2, ATP6V0A1, TGFBR3, HDAC9
- ## 2 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, EFNB2, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 3 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 4 DLC1, ENAH, NRP1, PGF, ZIC2, TGFB2, CD44, ILK, SEMA3C, RET, AR, RXRA, VANGL2, LEF1, TNNT2, HHEX, MIB1, NCOA3, FOXC2, FOXC1, TGFB1I1, WNT5A, COBL, BBS4, FGFR3, TNC, BMPR2, CTNND1, EGLN1, NR3C1, SOX9, TCF7L1, IGF1R, FOXQ1, MACF1, HOXA5, BCL2, PLXND1, CAR2, ACTC1, TBX4, SMAD3, FZD3, SHANK3, FZD6, HOXB4, FREM2, TSC2, ZIC5, TGFBR3, APAF1
- ## 5 GNA13, CAV1, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, CDH2, MEIS1, WT1, TGFB2, WARS, PTK2, CERKL, APOE, CXCR4, ANG, SEMA3C, PLCD1, NOS2, MKL2, PLXND1, FIGF, FGF2, PTPRJ, TGFBR2, TBX4, NF1, TBX1, TNNI3, PNPLA6, VASH1, THY1, NUS1, MEOX2, ECSCR, AMOT, HBEGF, FOXC2, FOXC1, HDAC7
- ## 6 CAV1, XIAP, AGFG1, ADORA2A, TNNC1, TBC1D9, LEPR, ABHD5, EDN1, ASAP2, ASAP3, SMAP1, TBC1D12, ANG, TBC1D14, MTCH1, TBC1D13, TBC1D4, TBC1D30, DHCR24, HIP1, VAV3, NOS1, NF1, MYH6, RICTOR, TBC1D22A, THY1, PLCE1, RNF7, NDEL1, CHML, IFT57, ACAP2, TSC2, ERN1, APAF1, ARAP3, ARAP2, ARAP1, HTR2A, F2R
- ## adj_pval
- ## 1 0.000002170
- ## 2 0.000010400
- ## 3 0.000007620
- ## 4 0.000119000
- ## 5 0.000720000
- ## 6 0.001171166
-
- #查看基因的数据格式
- head(EC$genelist)
-
- ## ID logFC AveExpr t P.Value adj.P.Val B
- ## 1 Slco1a4 6.645388 1.2168670 88.65515 1.32e-18 2.73e-14 29.02715
- ## 2 Slc19a3 6.281525 1.1600468 69.95094 2.41e-17 2.49e-13 27.62917
- ## 3 Ddc 4.483338 0.8365231 65.57836 5.31e-17 3.65e-13 27.18476
- ## 4 Slco1c1 6.469384 1.3558865 59.87613 1.62e-16 8.34e-13 26.51242
- ## 5 Sema3c 5.515630 2.3252117 58.53141 2.14e-16 8.81e-13 26.33626
- ## 6 Slc38a3 4.761755 0.9218670 54.11559 5.58e-16 1.76e-12 25.70308
Kun olet ymmärtänyt kaksi syöttötietomuotoa, voit käyttääcirlce_dattoiminto piirustustietojen luomiseen.
- # 生成画图所需的数据格式
- circ <- circle_dat(EC$david, EC$genelist)
-
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
circObjektissa on kahdeksan datasaraketta, nimittäin
Luokka: BP (biologinen prosessi), CC (Cellular Component) tai MF (Molecular Function)
ID: GO id (valinnainen sarake, jos haluat käyttää toiminnallista analyysityökalua, joka ei perustu GO id:hen, et voi valita ID-saraketta; ID voi tässä olla myös KEGG ID)
termi: GO polku
count: geenien lukumäärä kussakin reitissä
geeni: geenin nimi - logFC: kunkin geenin logFC-arvo
adj_pval: säädetty p-arvo, polkujen, joiden adj_pval <0,05, katsotaan olevan merkittävästi rikastunut
zscore: zscore ei viittaa tilastolliseen normalisointimenetelmään, vaan se on helposti laskettava arvo, jonka avulla voidaan arvioida, väheneekö biologinen prosessi (/molekyylifunktio/solukomponentti) todennäköisemmin (negatiivinen arvo) vai lisääntyykö (positiivinen arvo).Laskentamenetelmä on ylössäädeltyjen geenien lukumäärä vähennettynä alassäänneltyjen geenien lukumäärällä jaettuna kunkin reitin geenien lukumäärän neliöjuurella.
Kun tarkastelemme tietoja ensimmäisen kerran, haluamme näyttää kaaviosta mahdollisimman monta reittiä, ja haluamme myös löytää arvokkaita reittejä, joten tarvitsemme joitain parametreja tärkeyden arvioimiseksi. Pylväskaavioita käytetään usein kuvaamaan näytetietoja, joten voimme käyttää GOBar-toimintoa luodaksesi nopeasti hyvännäköisen pylväskaavion.
Ensin luodaan yksinkertainen pylväskaavio. Vaaka-akseli onGO Terms, heidän mukaansazscoreLajittele palkit; pystyakseli on-log(adj p-value);Väri edustaazscore, sininen osoittaaz-scoreon negatiivinen arvo, geenin ilmentyminen vastaavassa reitissä todennäköisemmin vähenee, merkitty punaisellaz-score on positiivinen arvo, geenin ilmentyminen vastaavassa reitissä lisääntyy todennäköisemmin. Järjestystä voidaan haluttaessa muuttaa asettamalla parametrin order.by.zscore arvoksi FALSE, jolloin palkit järjestetään niiden merkityksen mukaan.
- # 生成简单的条形图
- GOBar(subset(circ, category == 'BP'))
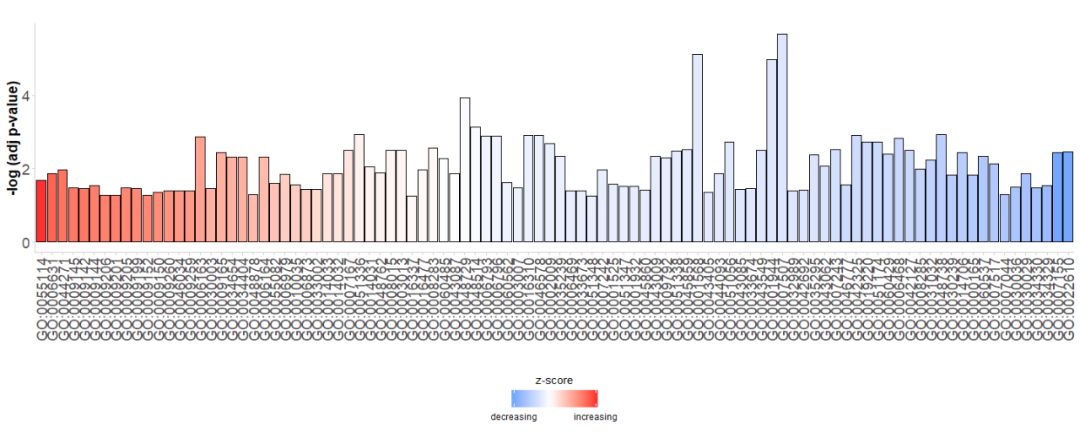
#GOBar(subset(circ, category == 'BP',order.by.zscore=FALSE))Lisäksi muuta näyttöparametria piirtääksesi pylväskaavion kanavan luokan mukaan.
- #根据通路的类别来绘制条形图
- GOBar(circ, display = 'multiple')
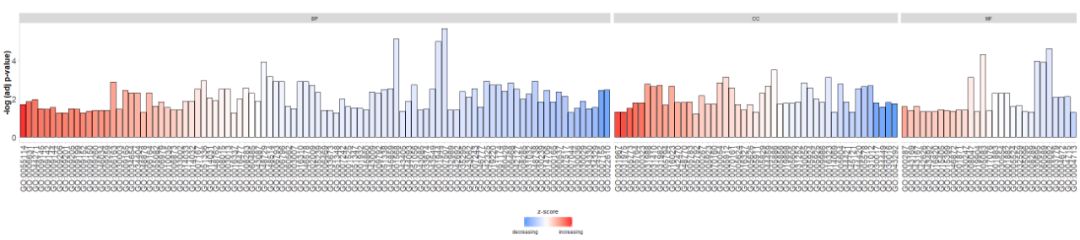
Lisää otsikko ja käytä parametrejazsc.colMuuttaazscores väri.
- # Facet the barplot, add a title and change the colour scale for the z-score
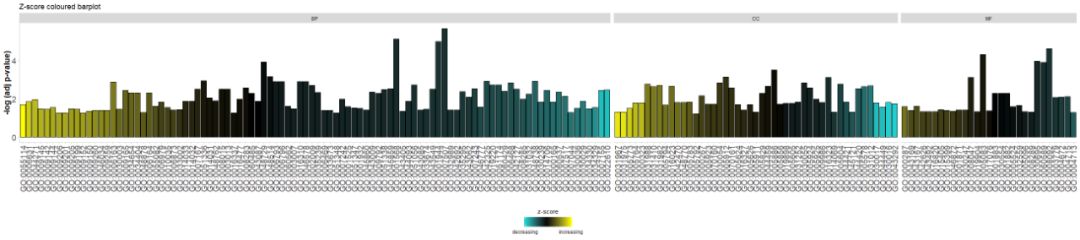
- GOBar(circ, display = 'multiple', title = 'Z-score coloured barplot', zsc.col = c('yellow', 'black', 'cyan'))
Pylväskaaviot ovat hyvin yleisiä ja helposti ymmärrettäviä, mutta voimme käyttää kuplakaavioita näyttääksemme lisätietoja tiedoista.
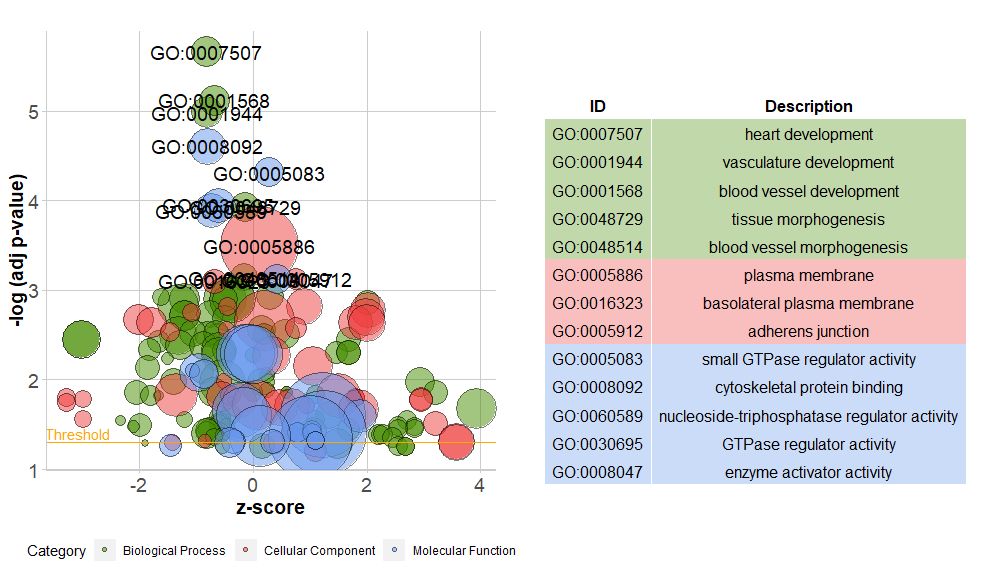
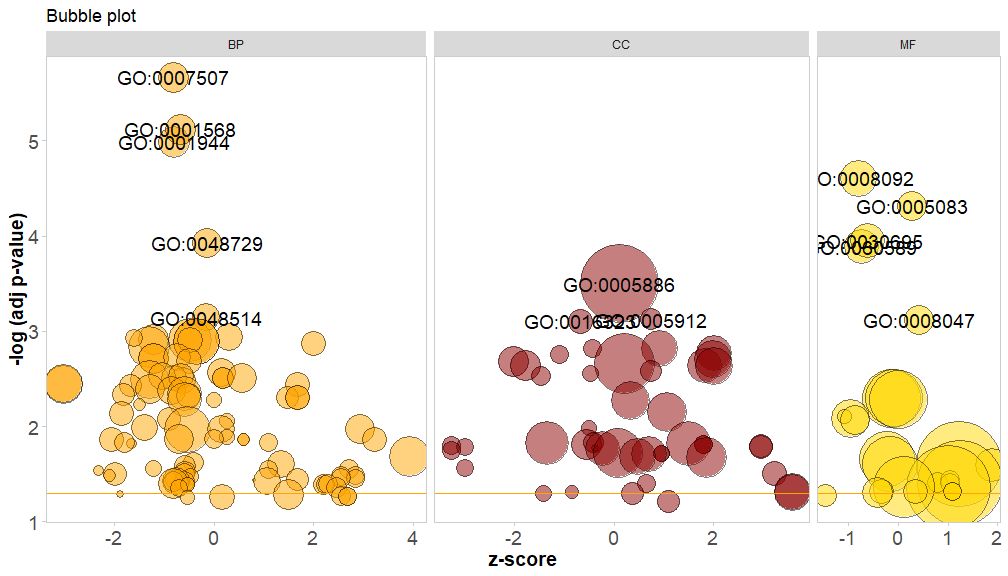
Vaaka-akseli onzscorePystyakseli on-log(adj p-value), kuten pylväskaavio, mitä korkeampi se on, sitä merkittävämpi ympyrän pinta-ala on suhteessa geenien määrään vastaavalla reitillä (;circ$count ); väri vastaa reittiä vastaavaa luokkaa, vihreä on biologinen prosessi, punainen on solukomponentti ja sininen on molekyylitoiminto.Voi tulla sisään?GOBubble Katso GOBubble-toiminnon ohjesivulta muuttaaksesi kaikkia kuvan parametreja. Oletusarvoisesti jokainen ympyrä on merkitty vastaavalla GO ID:llä, ja oikealla näkyy myös taulukko, joka näyttää vastaavan GO ID:n ja GO-termin välisen suhteen.Parametrit voidaan asettaatable.legendvartenFALSE piilottaaksesi sen. Jos haluat näyttää polun kuvauksen, aseta parametrin ID arvoksi FALSE.Rajallisen tilan ja päällekkäisten ympyröiden vuoksi kaikkia ympyröitä ei kuitenkaan ole merkitty, vain-log(adj p-value) > 3(oletus on 5).
- # 生成泡泡图,并展示-log(adj p-value) > 3 的通路的GO ID
- GOBubble(circ, labels = 3)
Jos haluat lisätä otsikon kuplakaavioon tai määrittää ympyrän värin ja näyttää kunkin luokan polut erikseen ja muuttaa näytettävää GO ID -kynnystä, voit lisätä seuraavat parametrit:
GOBubble(circ, title = 'Bubble plot', colour = c('orange', 'darkred', 'gold'), display = 'multiple', labels = 3)
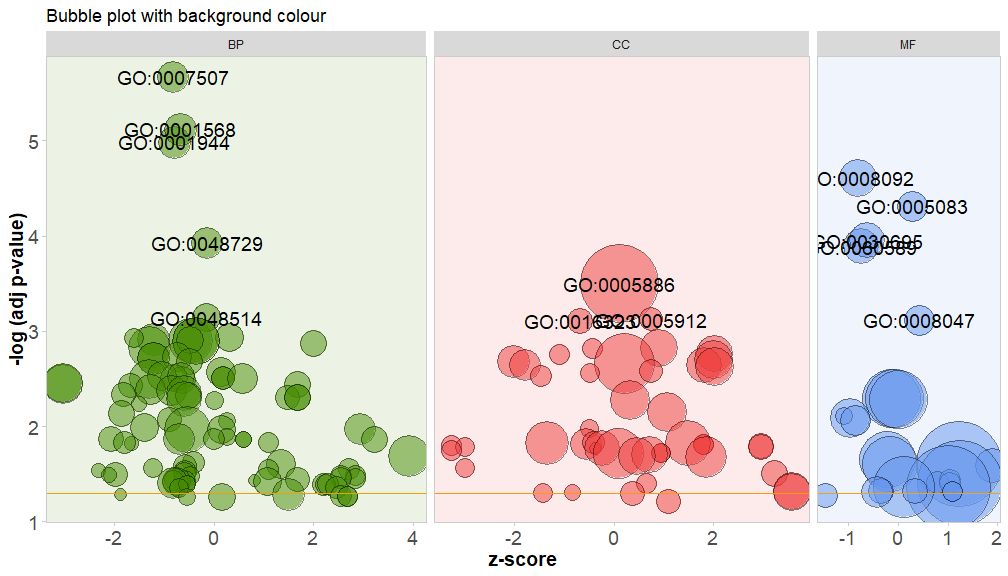
Väritä kanavan luokan tausta asettamalla parametrin bg.col arvoksi TOSI.
GOBubble(circ, title = 'Bubble plot with background colour', display = 'multiple', bg.col = T, labels = 3)
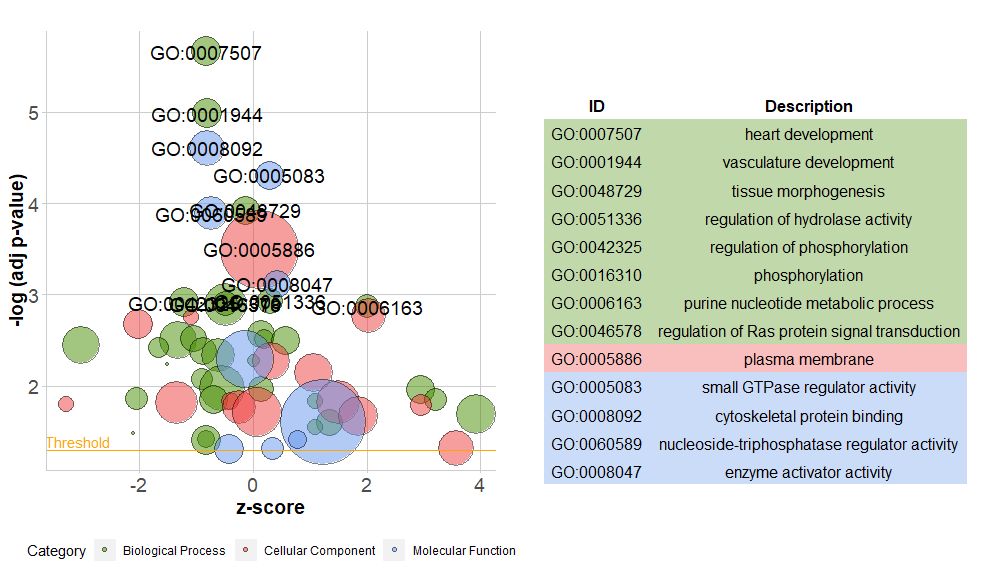
Paketin uusi versio sisältää uuden toiminnonreduce_overlap , tämä toiminto voi vähentää redundanttien kohteiden määrää, toisin sanoen se voi poistaa kaikki reitit, joiden geenien päällekkäisyys on suurempi tai yhtä suuri kuin asetettu kynnys, ja säilyttää vain yhden polun kustakin ryhmästä edustajana riippumatta siitä, näytetäänkö kaikki polkuja GO:ssa. Redundanttien termien määrää vähentämällä kaavioiden (kuten kuplakaavioiden) luettavuus paranee merkittävästi.
- # reduce_overlap,参数设置为0.75
- reduced_circ <- reduce_overlap(circ, overlap = 0.75)
-
- GOBubble(reduced_circ, labels = 2.8)
Vaikka kaikki tiedot näyttävä kaavio voi auttaa meitä selvittämään, mitkä reitit ovat merkityksellisimpiä, todellisuus riippuu silti hypoteeseista ja ideoista, jotka haluat vahvistaa tiedoilla, eivätkä tärkeimmät reitit välttämättä ole niitä, joista olet kiinnostunut. Siksi valittaessa manuaalisesti arvokasta reittijoukkoa (EC$process ), tarvitsemme kaavion, joka näyttää meille yksityiskohtaisempia tietoja tästä tietystä reittijoukosta.Mutta näiden lukujen esittäminen aiheuttaa ongelman: joskus niitä on vaikea tulkitazscore Tiedot toimitettu.Loppujen lopuksi tämä laskentamenetelmä ei ole universaali Kuten yllä näkyy, se on yksinkertaisesti ylössäädeltyjen geenien lukumäärä miinus alassäänneltyjen geenien lukumäärä jaettuna kunkin reitin geenien lukumäärän neliöjuurella.GOCircleTuloksena oleva kaavio korostaa myös tätä tosiasiaa.
Ympyräkaavion ulompi ympyrä näyttää kunkin reitin geenien logFC-arvon hajapisteinä. Punaiset ympyrät osoittavat ylössäätöä ja siniset alasäätelyä.Parametreja voidaan käyttäälfc.col Vaihda väri. Tämä selittää myös sen, miksi joissakin tapauksissa erittäin tärkeiden polkujen zs-pisteet ovat lähellä nollaa. Zscore nolla ei tarkoita, että kanava on merkityksetön. Se vain osoittaa, että zscore on karkea mitta, koska ilmeisesti zscore ei myöskään ota huomioon yksittäisten geenien toiminnallista tasoa ja aktivaatioriippuvuutta biologisissa prosesseissa.
GOCircle(circ)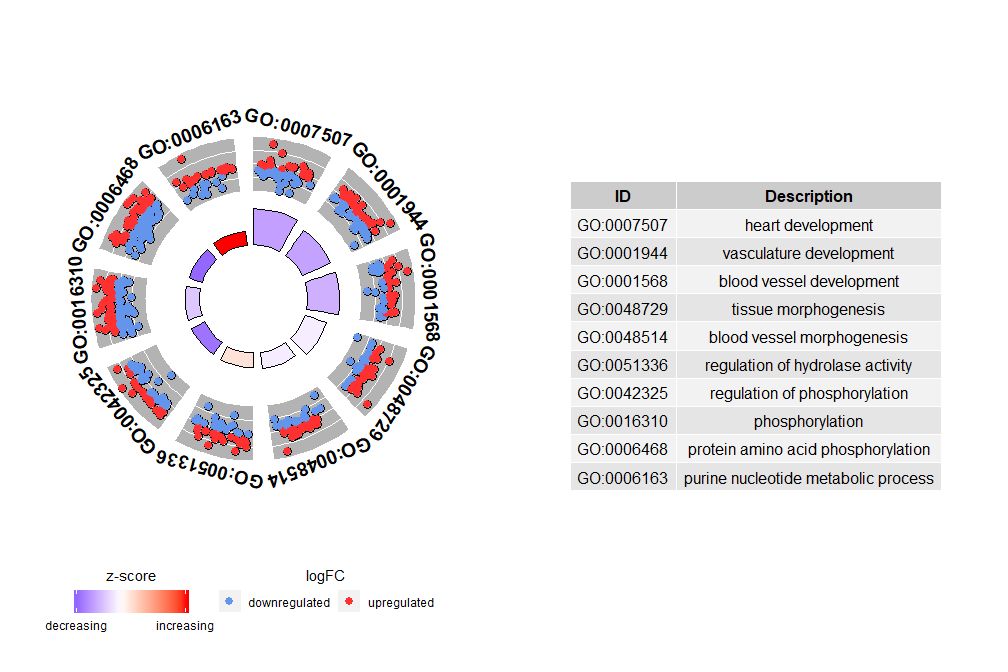
nsub Parametrit voivat olla numeroita tai merkkivektoreita. Jos se on merkkivektori, se sisältää GO ID:n tai näytettävän polun;
- # 生成特定通路的圈图
- IDs <- c('GO:0007507', 'GO:0001568', 'GO:0001944', 'GO:0048729', 'GO:0048514', 'GO:0005886', 'GO:0008092', 'GO:0008047')
- GOCircle(circ, nsub = IDs)
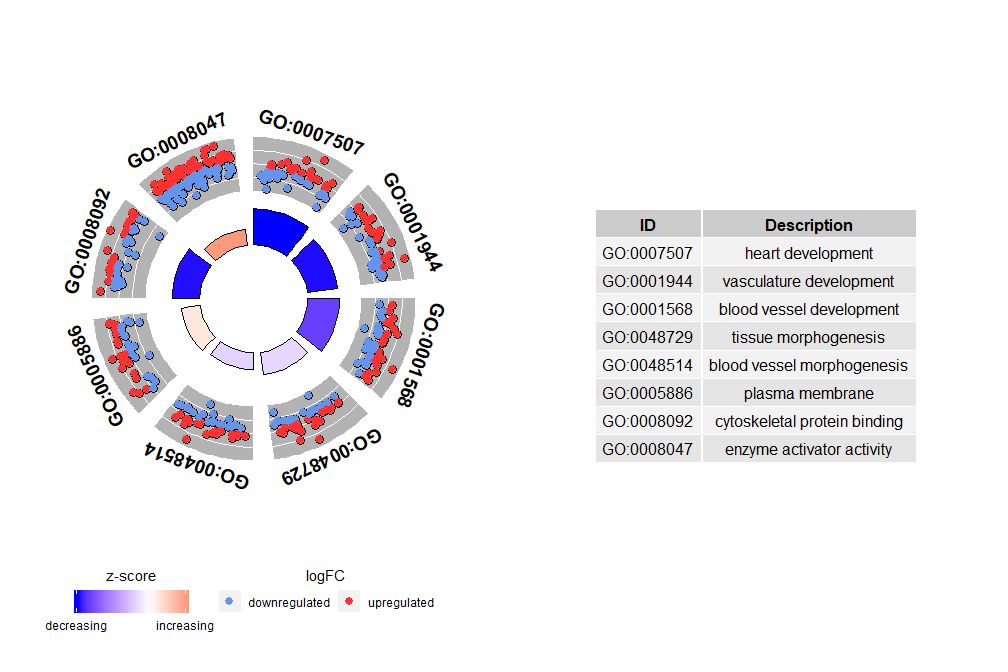
Jos nsub on numeerinen vektori, numero määrittää näytettävän numeron. Se alkaa syötetietokehyksen ensimmäiseltä riviltä. Tämä visualisointi toimii vain pienemmillä tiedoilla. Kanavien enimmäismäärä on oletuksena 12. Vaikka kanavien määrä vähenee, näytettävän tiedon määrä kasvaa.
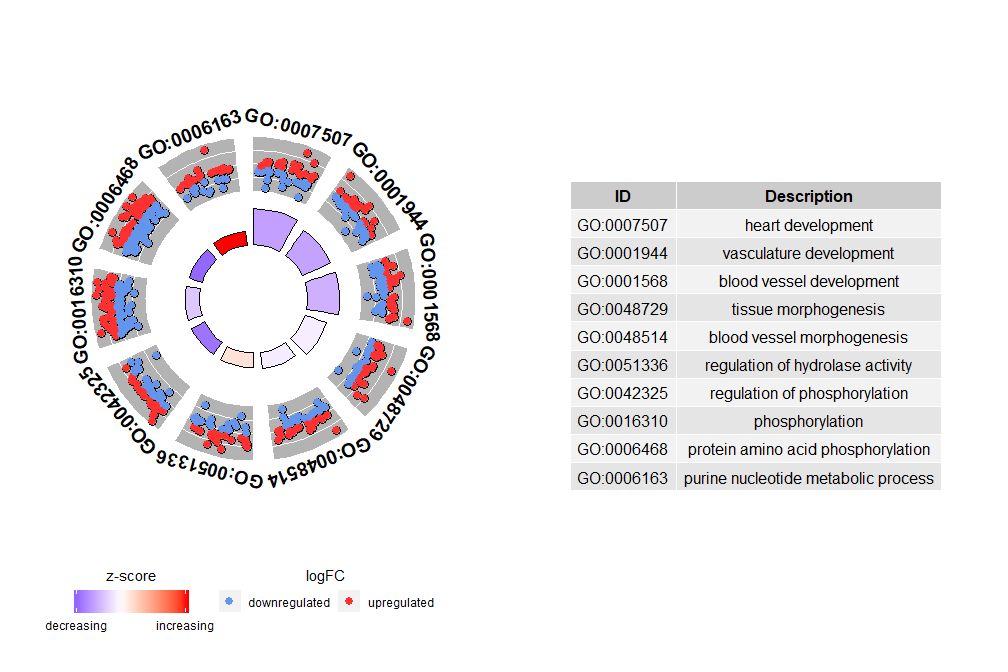
- # 圈图展示数据前十个通路
- GOCircle(circ, nsub = 10)
GOChord voi näyttää valittujen geenien ja polkujen välisen suhteen ja geenien logFC:n.Ensin sinun on syötettävä matriisi, jonka voit rakentaa itse0-1Matrix, voit myös käyttää toimintojachord_dat Rakentaa. Tällä funktiolla on kolme parametria: data, geenit ja prosessi, joista kahdella viimeisellä parametrilla on oltava vähintään yksi parametri.Sitten funktiocircle_datYhdistä ilmentymistiedot toiminnallisten analyysien tuloksiin.
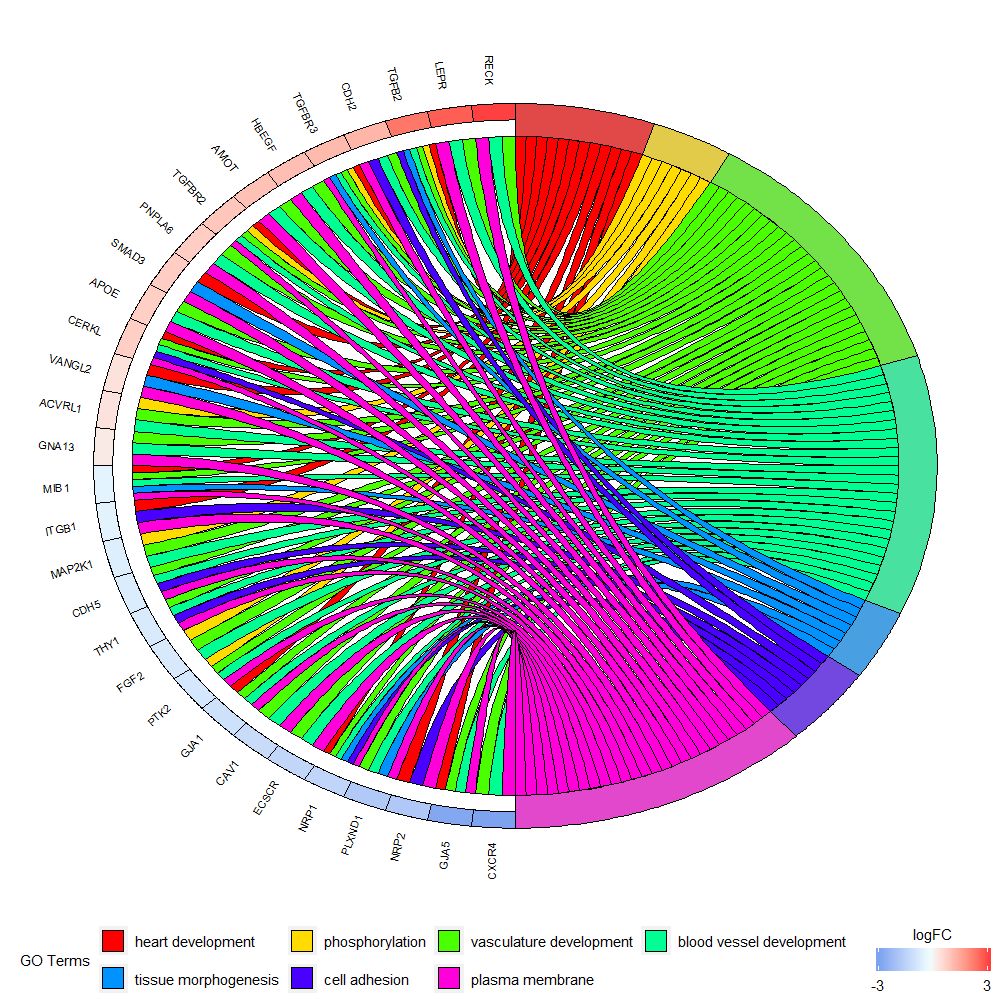
Pylväskaaviot ja kuplakaaviot voivat antaa sinulle ensivaikutelman tiedoista. Nyt voit valita joitain geenejä ja polkuja, jotka ovat mielestämme arvokkaita. Vaikka GOCircle lisää kerroksen geenien ilmentymisarvon näyttämiseen reiteillä, siitä puuttuu yksittäisiä tietoja geenien ja useiden reittien väliset suhteet. Ei ole helppoa selvittää, liittyvätkö tietyt geenit useisiin prosesseihin. GOChord korvaa GOCirclen puutteet. Luotujen tietojen rivit ovat geenejä ja sarakkeet ovat polkuja.
- # 找到感兴趣的的基因,这里我们以EC$genes为例
- head(EC$genes)
-
- ## ID logFC
- ## 1 PTK2 -0.6527904
- ## 2 GNA13 0.3711599
- ## 3 LEPR 2.6539788
- ## 4 APOE 0.8698346
- ## 5 CXCR4 -2.5647537
- ## 6 RECK 3.6926860
-
- # 获得感兴趣基因的通路
- EC$process
-
- ## [1] "heart development" "phosphorylation"
- ## [3] "vasculature development" "blood vessel development"
- ## [5] "tissue morphogenesis" "cell adhesion"
- ## [7] "plasma membrane"
-
- # 使用chord_dat构建矩阵
- chord <- chord_dat(circ, EC$genes, EC$process)
- head(chord)
-
- ## heart development phosphorylation vasculature development
- ## PTK2 0 1 1
- ## GNA13 0 0 1
- ## LEPR 0 0 1
- ## APOE 0 0 1
- ## CXCR4 0 0 1
- ## RECK 0 0 1
- ## blood vessel development tissue morphogenesis cell adhesion
- ## PTK2 1 0 0
- ## GNA13 1 0 0
- ## LEPR 1 0 0
- ## APOE 1 0 0
- ## CXCR4 1 0 0
- ## RECK 1 0 0
- ## plasma membrane logFC
- ## PTK2 1 -0.6527904
- ## GNA13 1 0.3711599
- ## LEPR 1 2.6539788
- ## APOE 1 0.8698346
- ## CXCR4 1 -2.5647537
- ## RECK 1 3.6926860
Esimerkissä välitettiin kaksi parametria. Jos vain geeniparametri on määritetty, tuloksena on luettelo valituista geeneistä ja kaikista prosessirakenteista, joissa on vähintään yksi määritetty geeni.0-1matriisi, jos vain määritettyprocessparametrit, tuloksena on, että kaikki geenit generoivat0-1 Vähintään yhdelle luettelon prosessille määritettyjen geenien matriisi. Huomaa, että vain geenien ja prosessiparametrien määrittäminen voi johtaa erittäin suureen 0-1-matriisiin, mikä johtaa hämmentävään visualisointitulokseen.
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
-
- # Generate the matrix with a list of selected genes
- chord_genes <- chord_dat(data = circ, genes = EC$genes)
- head(chord_genes)
-
- ## heart development vasculature development blood vessel development
- ## PTK2 0 1 1
- ## GNA13 0 1 1
- ## LEPR 0 1 1
- ## APOE 0 1 1
- ## CXCR4 0 1 1
Tämä kaavio on tarkoitettu näyttämään pienempi osa suuriulotteisia tietoja. Pääsääntöisesti kaksi parametria voidaan säätää:gene.orderjanlfc . Geeniparametriksi voidaan määrittää 'logFC', 'aakkosellinen', 'ei mitään'. Itse asiassa määritämme yleensä geeniparametrin logFC:ksi, nlfc-parametri on yksi tämän toiminnon tärkeimmistä parametreista, koska se pystyy käsittelemään sitä, kuinka kullakin geenillä on 0 tai enemmän logFC-arvoja matriisissa. Siksi meidän tulisi määrittää parametrit virheiden välttämiseksi.
Jos sinulla on esimerkiksi matriisi ilman logFC-arvoja, sinun on asetettavanlfc=0 Tai suorita differentiaalinen ilmentymisanalyysi geeneille useissa olosuhteissa tai erissä. Tässä tapauksessa jokainen geeni sisältää useita logFC-arvoja, ja nlfc=logFC-sarakkeen numero on asetettava. Oletusarvo on "1", koska uskotaan, että suurimman osan ajasta geeniä kohden on vain yksi logFC-arvo. Käytä space-parametria määrittääksesi tilan logFC:tä edustavien värillisten suorakulmioiden välillä. Parametri gene.size määrittää geenin nimen kirjasinkoon ja gene.space määrittää geeninimien välisen tilan.
- chord <- chord_dat(data = circ, genes = EC$genes, process = EC$process)
- GOChord(chord, space = 0.02, gene.order = 'logFC', gene.space = 0.25, gene.size = 5)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
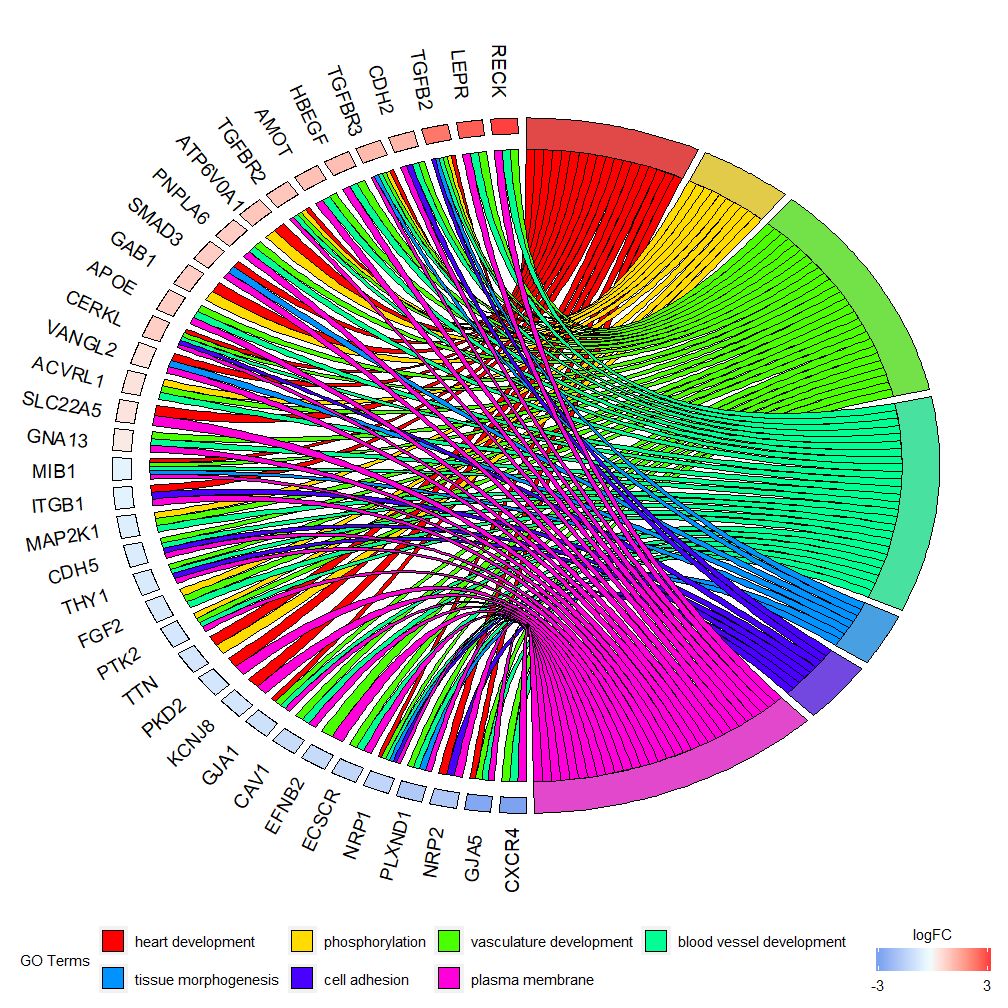
Voidaan asettaa logFC-arvon mukaangene.order=‘logFC’ , lajittele geenit logFC-arvojensa mukaan. Joskus kuvasta voi tulla hieman tungosta ja tämä voidaan automatisoida käyttämällä limit-parametria vähentämään näytettävien geenien tai polkujen määrää. Raja on vektori, jossa on kaksi raja-arvoa (oletus on c(0,0)). Ensimmäinen arvo määrittää vähimmäismäärän polkuja, joihin geeni on osoitettava. Toinen arvo määrittää polulle osoitettujen geenien lukumäärän.
- # 仅显示分配给至少三个通路的基因
- GOChord(chord, limit = c(3, 0), gene.order = 'logFC')
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
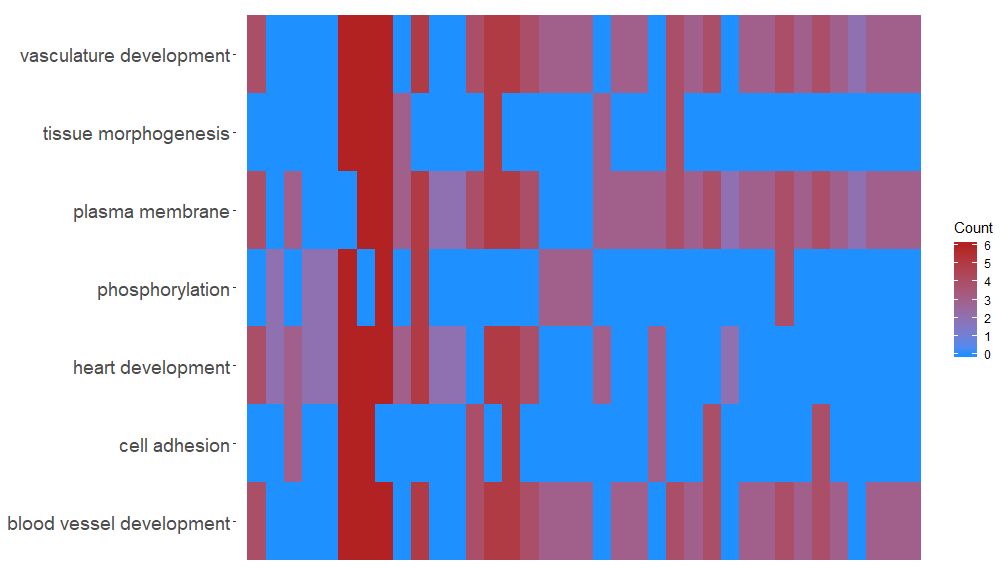
GOHeat-funktio voi näyttää geenien ja polkujen välisen suhteen lämpökartan avulla, joka on samanlainen kuin GOChord. Biologiset prosessit näytetään vaakasuunnassa ja geenit pystysuorassa. Jokainen sarake on jaettu pieniin suorakulmioihin, ja väri riippuu yleensä logFC-arvosta. Lisäksi samanlaisilla toiminnallisilla reiteillä rikastuneet geenit ryhmiteltiin. Lämpökartan värin valintaan on kaksi tilaa nlfc-parametreista riippuen. Jos nlfc = 0, väri on kunkin geenin rikastettujen reittien lukumäärä. Katso lisätietoja esimerkeistä:
- # First, we use the chord object without logFC column to create the heatmap
- GOHeat(chord[,-8], nlfc = 0)
GOHeat(chord[,-8])Väri vastaa geenin logFC:tä, jos nlfc = 1
GOHeat(chord, nlfc = 1, fill.col = c('red', 'yellow', 'green'))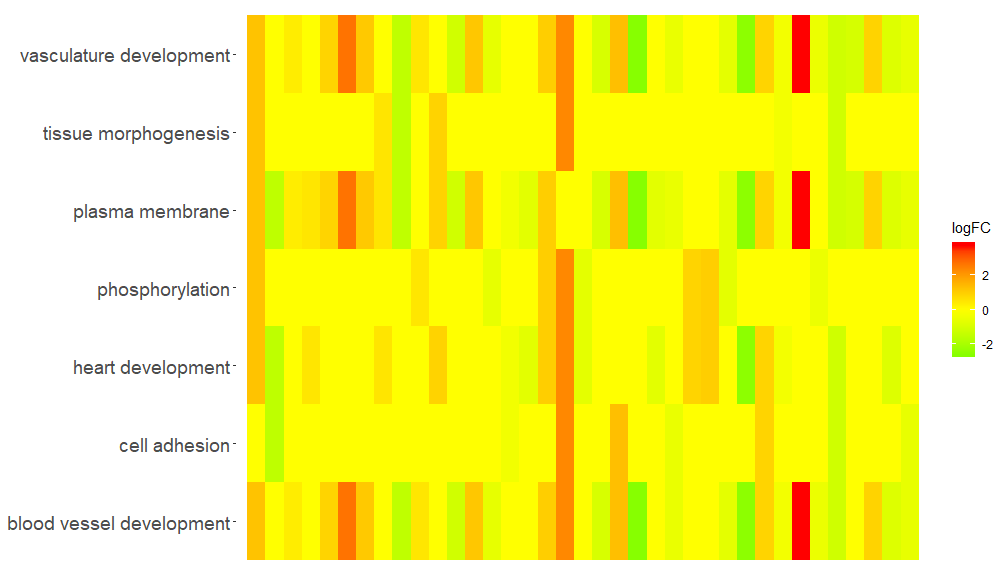
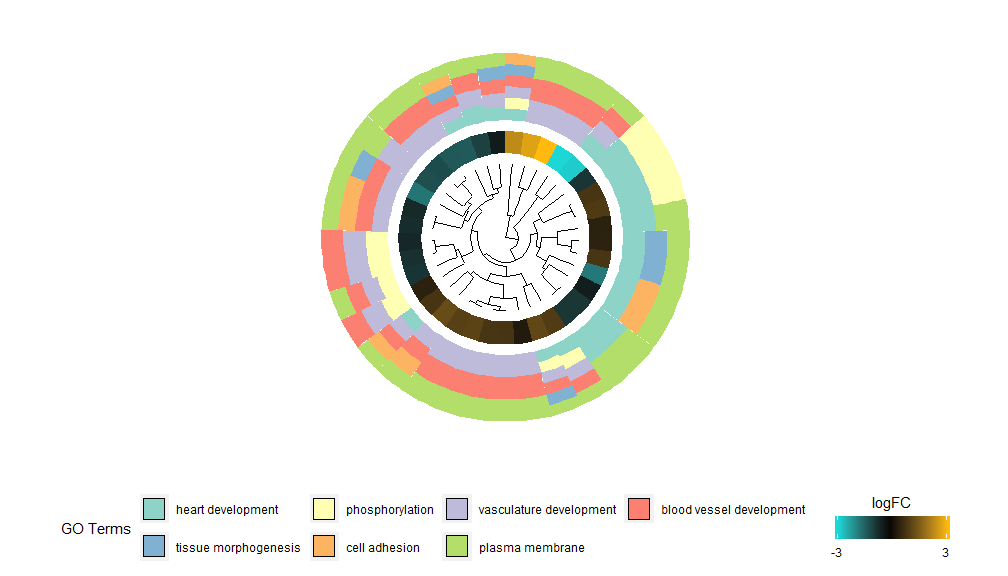
GOCluster-toiminnallisuuden ideana on näyttää mahdollisimman paljon tietoa. Tässä on esimerkki:
- GOCluster(circ, EC$process, clust.by = 'logFC', term.width = 2)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
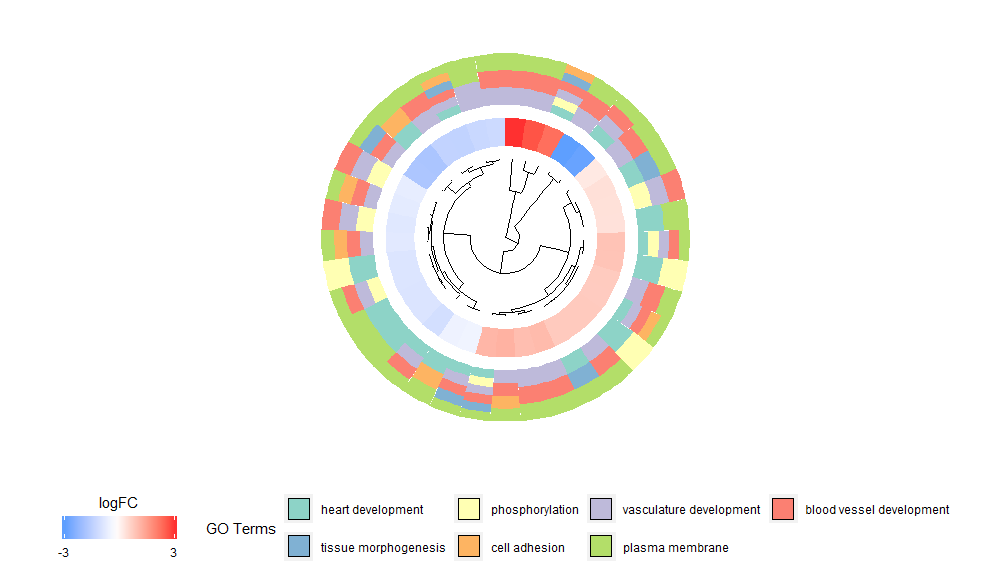
Hierarkkinen klusterointi on suosittu valvomaton klusterointianalyysimenetelmä geenien ilmentymiseen, joka varmistaa geenien puolueettoman ryhmittelyn ekspressiomallin mukaan, joten klusterit, jotka klusteroituvat, voivat sisältää useita yhteissäänneltyjen tai toiminnallisesti sukua olevien geenien ryhmiä. GOCluster käyttäähclust Menetelmä suorittaa geeniekspressioprofiilien hierarkkisen klusteroinnin. Jos haluat muuttaa etäisyysmetriikkaa tai klusterointialgoritmia, käytä parametreja metriikka ja klusteri. Tuloksena oleva dendrogrammi voidaan muuntaa ggdendron avulla ja visualisoida ggplot2:lla. Valitse pyöreä asettelu, koska se ei ole vain tehokas, vaan myös visuaalisesti houkutteleva. Ensimmäinen ympyrä dendrogrammin vieressä edustaa geenin logFC:tä, joka on itse asiassa klusteripuun lehti. Jos olet kiinnostunut useista kontrasteista, voit muokata nlfc-parametria, oletuksena se on "1", joten vain yksi rengas piirretään. LogFC-arvot on värikoodattu käyttämällä käyttäjän määriteltävää väriasteikkoa (lfc.col), joka edustaa geenille määritettyä reittiä. Hyvältä näyttämiseksi on kanavien määrää vähennetty ja kanavien väriä voidaan muuttaa parametrilla term.col.yhä saatavilla?GOCluster nähdäksesi kuinka parametreja muutetaan. Tämän funktion tärkein parametri on cluster.by, joka voidaan määrittää klusteriksi geenin ilmentymismallien ('logFC', kuten yllä on esitetty) tai funktionaalisten luokkien ('terms') mukaan.
- GOCluster(circ, EC$process, clust.by = 'term', lfc.col = c('darkgoldenrod1', 'black', 'cyan1'))
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
Venn-diagrammeja voidaan käyttää erilaisten erilaisesti ilmentyneiden geenien luetteloiden välisten suhteiden havaitsemiseen tai useiden reittien geenien leikkauspisteiden tutkimiseen toiminnallisissa analyyseissä. Venn-kaaviot eivät näytä vain päällekkäisten geenien määrää, vaan myös tietoa geenin ilmentymiskuviosta (yleensä ylös-, usein alas- tai vastasäädelty). Tällä hetkellä syötteenä käytetään enintään kolmea tietojoukkoa. Syöttötieto sisältää vähintään kaksi saraketta: yksi geenien nimille ja yksi logFC-arvoille.
- l1 <- subset(circ, term == 'heart development', c(genes,logFC))
- l2 <- subset(circ, term == 'plasma membrane', c(genes,logFC))
- l3 <- subset(circ, term == 'tissue morphogenesis', c(genes,logFC))
- GOVenn(l1,l2,l3, label = c('heart development', 'plasma membrane', 'tissue morphogenesis'))
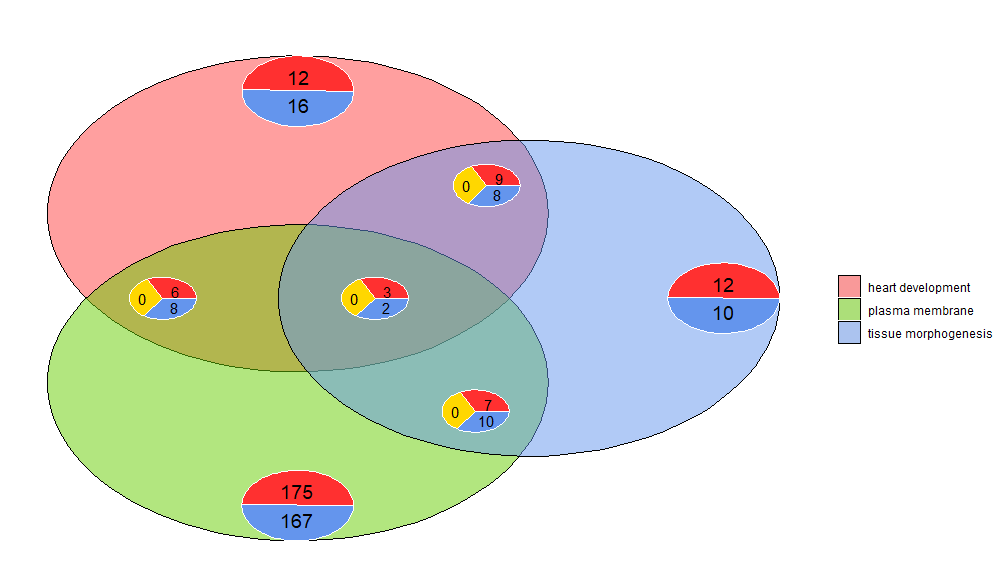
Esimerkiksi sydämen kehityksessä ja kudosten morfogeneesissä on 22 geeniä, joista 12 on säädelty ylöspäin ja 10 alasäädelty. Tärkeä asia on huomata, että ympyräkaaviot eivät näytä tarpeettomia tietoja. Siksi, jos kolmea tietojoukkoa verrataan, kaikille tietojoukoille yhteiset geenit (keskimmäinen ympyräkaavio) eivät sisälly muihin ympyräkaavioihin. Tämä työkalu on saatavilla osoitteessa shinyapp https://wwalter.shinyapps.io/Venn/, verkkotyökalu on interaktiivisempi, ympyrän pinta-ala on verrannollinen tietojoukon geenien määrään ja liukusäätimellä voidaan siirtää pieni ympyräkaavio, ja siinä on GOVenn-ominaisuudet kaikki vaihtoehdot juonen asettelun muuttamiseen sekä kuvien ja geeniluetteloiden lataamiseen.
Ohjelmiston kotisivu: https://wencke.github.io/

Hän on omistautunut teknologian tutkimukselle yli 30 vuoden ajan ja hallitsee useita kieliä, kuten java, linux, javascript, php, css jne. Hän on tehnyt paljon työtä avoimen lähdekoodin alalla Kehittäjän dokumentaatioasema jakaaksesi joitain teknologian kehityksen ongelmia tulevaa käyttöä varten

