
le mie informazioni di contatto
Posta[email protected]
2024-07-12
한어Русский языкEnglishFrançaisIndonesianSanskrit日本語DeutschPortuguêsΕλληνικάespañolItalianoSuomalainenLatina
Il pacchetto GOPlot viene utilizzato per la visualizzazione di dati biologici. Piuttosto, questo pacchetto integra e visualizza i dati di espressione con i risultati delle analisi funzionali.Ma fa attenzioneQuesto pacchetto non può essere utilizzato per eseguire queste analisi, ma solo per visualizzare i risultati. . In tutti i campi della scienza, è difficile descrivere realisticamente le cose a causa dei vincoli di spazio e della semplicità richiesta per i risultati, quindi è necessario visualizzare le informazioni e utilizzare immagini per trasmetterle. Una grafica ben progettata fornisce più informazioni in meno spazio. L'idea del pacchetto è quella di consentire agli utenti di esaminare rapidamente grandi quantità di dati, rivelare tendenze nei dati e trovare modelli e correlazioni nei dati.
La visualizzazione dei dati può aiutarci a trovare risposte a domande biologiche, giudicare una determinata ipotesi e persino scoprire diverse angolazioni per indagare su diversi problemi. E le funzioni di tracciamento di questo pacchetto sono sviluppate in base alla struttura gerarchica dei dati, iniziando con i dati complessivi e terminando con un sottoinsieme di geni selezionati e percorsi corrispondenti.
Spieghiamolo concretamente con un esempio.
Chiamiamo i dati forniti con GOplot, che provengono da GEOGSE47067, contenente informazioni sul trascrittoma delle cellule endoteliali di due tessuti (cervello e cuore). Per ulteriori informazioni, vedere l'articolo di Nolan et al https://www.ncbi.nlm.nih.gov/pubmed/23871589, e poi.I dati vengono normalizzati e vengono trovati i geni espressi in modo differenziale., quindi utilizzare lo strumento di annotazione della funzione DAVID (i dati di annotazione della funzione DAVID vengono aggiornati lentamente e non è consigliato ora. Si consiglia di utilizzareGo East, il miglior strumento online di analisi dell'arricchimento GOEQuesto sito web, che può eseguire analisi di arricchimento in un solo passaggio, è stato citato più di 350 volte da CNS e altri prima della sua pubblicazione.Eseguire analisi di arricchimento,Master GSEA in un articolo, tutorial super dettagliato) Annotazione genica di geni differenzialmente espressi (adjusted p-value < 0.05 ) e analisi di arricchimento funzionale. Questo set di dati contiene le seguenti cinque categorie di dati:
| nome | descrivere | Dimensioni del set di dati |
|---|---|---|
| EC$set | Espressione genica normalizzata nelle cellule endoteliali del cervello e del cuore (3 repliche) | 20644 x 7 |
| EC$genelist | Geni espressi in modo differenziale (valore p aggiustato <0,05) | 2039 x 7 |
| EC$david | Risultati dell'analisi di arricchimento funzionale di geni differenziali utilizzando DAVID | 174 x 5 |
| EC$gene | Geni e logFC | 37x2 |
| EC$process | Vettori di caratteristiche selezionati per processi biologici arricchiti | 7 |
Vogliamo vedere i percorsi arricchiti del GO di geni espressi in modo differenziale, ma prima di iniziare a disegnare dobbiamo fornire dati che soddisfino i requisiti di formato.In generale, i dati necessari per disegnare il grafico li fornisci tu, maC'è una funzione in questo pacchettocircle_datPuò aiutarci a gestire il formato dei dati。circle_datPuò combinare i risultati dell'analisi di arricchimento funzionale di geni selezionati e i loro valori logFC, principalmente per geni espressi in modo differenziale.circle_dat L'utilizzo è molto semplice, basta leggere due dati. I primi dati contengono i risultati dell'analisi dell'arricchimento funzionale, con almeno quattro colonne (categoria dell'analisi dell'arricchimento funzionale, percorso, gene, valore p corretto).Il secondo dato riguarda il gene selezionato e il suo logFC, questi dati possono essere la fontelimmaI risultati dell'analisi statistica (Nota dalle biografie: assicurarsi di prestare attenzione a due fileCome vengono chiamati i geniSii coerente, come tuttiGene symbol ). Diamo un'occhiata ai formati di dati menzionati sopra con esempi.
- #安装已发布的稳定版本
- #install.packages('GOplot')
- #安装github上的开发版本
- #install_github('wencke/wencke.github.io')
- #载入包
- library(GOplot)
- #读入包内自带的数据
- data(EC)
- #查看功能富集分析结果的数据格式
- head(EC$david)
-
- ## Category ID Term
- ## 1 BP GO:0007507 heart development
- ## 2 BP GO:0001944 vasculature development
- ## 3 BP GO:0001568 blood vessel development
- ## 4 BP GO:0048729 tissue morphogenesis
- ## 5 BP GO:0048514 blood vessel morphogenesis
- ## 6 BP GO:0051336 regulation of hydrolase activity
- ## Genes
- ## 1 DLC1, NRP2, NRP1, EDN1, PDLIM3, GJA1, TTN, GJA5, ZIC3, TGFB2, CERKL, GATA6, COL4A3BP, GAB1, SEMA3C, MKL2, SLC22A5, MB, PTPRJ, RXRA, VANGL2, MYH6, TNNT2, HHEX, MURC, MIB1, FOXC2, FOXC1, ADAM19, MYL2, TCAP, EGLN1, SOX9, ITGB1, CHD7, HEXIM1, PKD2, NFATC4, PCSK5, ACTC1, TGFBR2, NF1, HSPG2, SMAD3, TBX1, TNNI3, CSRP3, FOXP1, KCNJ8, PLN, TSC2, ATP6V0A1, TGFBR3, HDAC9
- ## 2 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, EFNB2, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 3 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 4 DLC1, ENAH, NRP1, PGF, ZIC2, TGFB2, CD44, ILK, SEMA3C, RET, AR, RXRA, VANGL2, LEF1, TNNT2, HHEX, MIB1, NCOA3, FOXC2, FOXC1, TGFB1I1, WNT5A, COBL, BBS4, FGFR3, TNC, BMPR2, CTNND1, EGLN1, NR3C1, SOX9, TCF7L1, IGF1R, FOXQ1, MACF1, HOXA5, BCL2, PLXND1, CAR2, ACTC1, TBX4, SMAD3, FZD3, SHANK3, FZD6, HOXB4, FREM2, TSC2, ZIC5, TGFBR3, APAF1
- ## 5 GNA13, CAV1, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, CDH2, MEIS1, WT1, TGFB2, WARS, PTK2, CERKL, APOE, CXCR4, ANG, SEMA3C, PLCD1, NOS2, MKL2, PLXND1, FIGF, FGF2, PTPRJ, TGFBR2, TBX4, NF1, TBX1, TNNI3, PNPLA6, VASH1, THY1, NUS1, MEOX2, ECSCR, AMOT, HBEGF, FOXC2, FOXC1, HDAC7
- ## 6 CAV1, XIAP, AGFG1, ADORA2A, TNNC1, TBC1D9, LEPR, ABHD5, EDN1, ASAP2, ASAP3, SMAP1, TBC1D12, ANG, TBC1D14, MTCH1, TBC1D13, TBC1D4, TBC1D30, DHCR24, HIP1, VAV3, NOS1, NF1, MYH6, RICTOR, TBC1D22A, THY1, PLCE1, RNF7, NDEL1, CHML, IFT57, ACAP2, TSC2, ERN1, APAF1, ARAP3, ARAP2, ARAP1, HTR2A, F2R
- ## adj_pval
- ## 1 0.000002170
- ## 2 0.000010400
- ## 3 0.000007620
- ## 4 0.000119000
- ## 5 0.000720000
- ## 6 0.001171166
-
- #查看基因的数据格式
- head(EC$genelist)
-
- ## ID logFC AveExpr t P.Value adj.P.Val B
- ## 1 Slco1a4 6.645388 1.2168670 88.65515 1.32e-18 2.73e-14 29.02715
- ## 2 Slc19a3 6.281525 1.1600468 69.95094 2.41e-17 2.49e-13 27.62917
- ## 3 Ddc 4.483338 0.8365231 65.57836 5.31e-17 3.65e-13 27.18476
- ## 4 Slco1c1 6.469384 1.3558865 59.87613 1.62e-16 8.34e-13 26.51242
- ## 5 Sema3c 5.515630 2.3252117 58.53141 2.14e-16 8.81e-13 26.33626
- ## 6 Slc38a3 4.761755 0.9218670 54.11559 5.58e-16 1.76e-12 25.70308
Dopo aver compreso i due formati di dati di input, è possibile utilizzarecirlce_datfunzione per generare dati di disegno.
- # 生成画图所需的数据格式
- circ <- circle_dat(EC$david, EC$genelist)
-
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
circL'oggetto ha otto colonne di dati, vale a dire
categoria: BP (processo biologico), CC (componente cellulare) o MF (funzione molecolare)
ID: GO id (colonna facoltativa, se si desidera utilizzare uno strumento di analisi funzionale che non è basato su GO id, non è possibile selezionare la colonna ID; l'ID qui può anche essere KEGG ID)
termine: percorso GO
conteggio: numero di geni in ciascuna via
gene: nome del gene - logFC: valore logFC di ciascun gene
adj_pval: valore p aggiustato, i percorsi con adj_pval<0,05 sono considerati significativamente arricchiti
zscore: zscore non si riferisce ad un metodo di normalizzazione statistica, ma è un valore facilmente calcolabile per stimare se un processo biologico (/funzione molecolare/componente cellulare) ha maggiori probabilità di diminuire (valore negativo) o aumentare (valore positivo).Il metodo di calcolo è il numero di geni sovraregolati meno il numero di geni sottoregolati diviso per la radice quadrata del numero di geni in ciascun percorso.
Quando esaminiamo per la prima volta i dati, vogliamo mostrare il maggior numero possibile di percorsi dal grafico e vogliamo anche trovare percorsi preziosi, quindi abbiamo bisogno di alcuni parametri per valutarne l'importanza. I grafici a barre vengono spesso utilizzati per descrivere dati di esempio, quindi possiamo utilizzare la funzione GOBar per creare rapidamente un grafico a barre di bell'aspetto.
Innanzitutto, viene generato direttamente un semplice grafico a barre. L'asse orizzontale èGO Terms, secondo lorozscoreOrdina le barre; l'asse verticale è-log(adj p-value);Il colore rappresentazscore, il blu indicaz-scoreè un valore negativo, è più probabile che l'espressione genica nel percorso corrispondente diminuisca, indicata in rossoz-score è un valore positivo, è più probabile che l'espressione genica nella via corrispondente aumenti. Se lo si desidera, l'ordine può essere modificato impostando il parametro order.by.zscore su FALSE, nel qual caso le barre vengono ordinate in base al loro significato.
- # 生成简单的条形图
- GOBar(subset(circ, category == 'BP'))
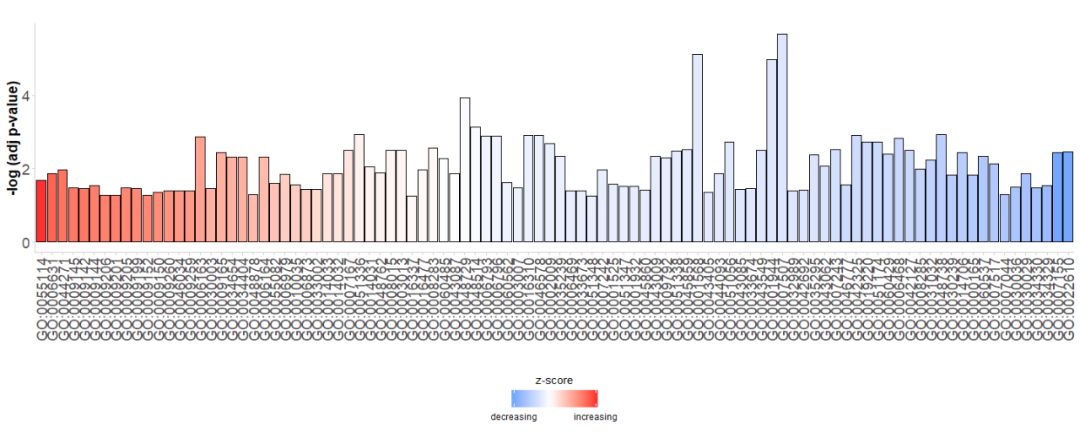
#GOBar(subset(circ, category == 'BP',order.by.zscore=FALSE))Inoltre, modificare il parametro di visualizzazione per disegnare un grafico a barre in base alla categoria del canale.
- #根据通路的类别来绘制条形图
- GOBar(circ, display = 'multiple')
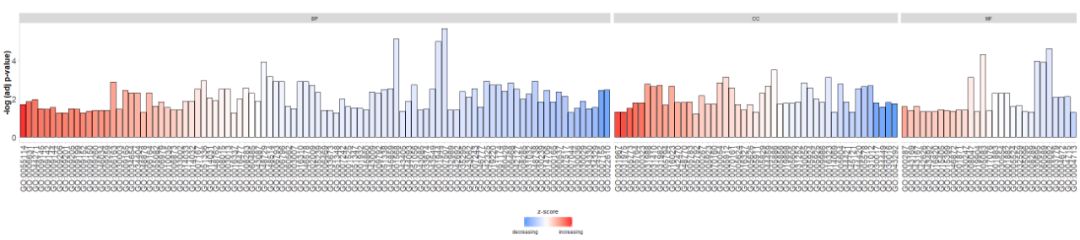
Aggiungi un titolo e utilizza i parametrizsc.colModificazscoreè il colore.
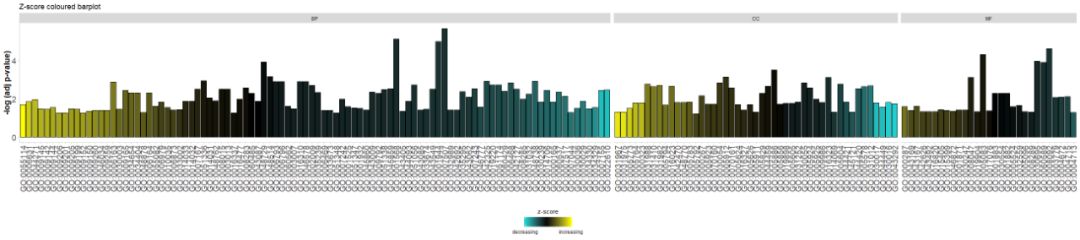
- # Facet the barplot, add a title and change the colour scale for the z-score
- GOBar(circ, display = 'multiple', title = 'Z-score coloured barplot', zsc.col = c('yellow', 'black', 'cyan'))
I grafici a barre sono molto comuni e facili da capire, ma possiamo utilizzare i grafici a bolle per visualizzare maggiori informazioni sui dati.
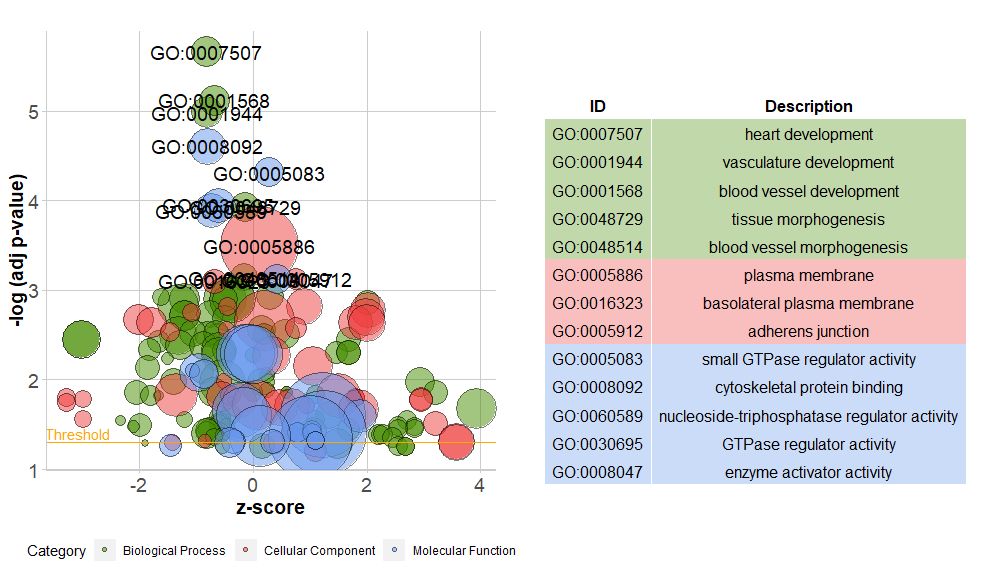
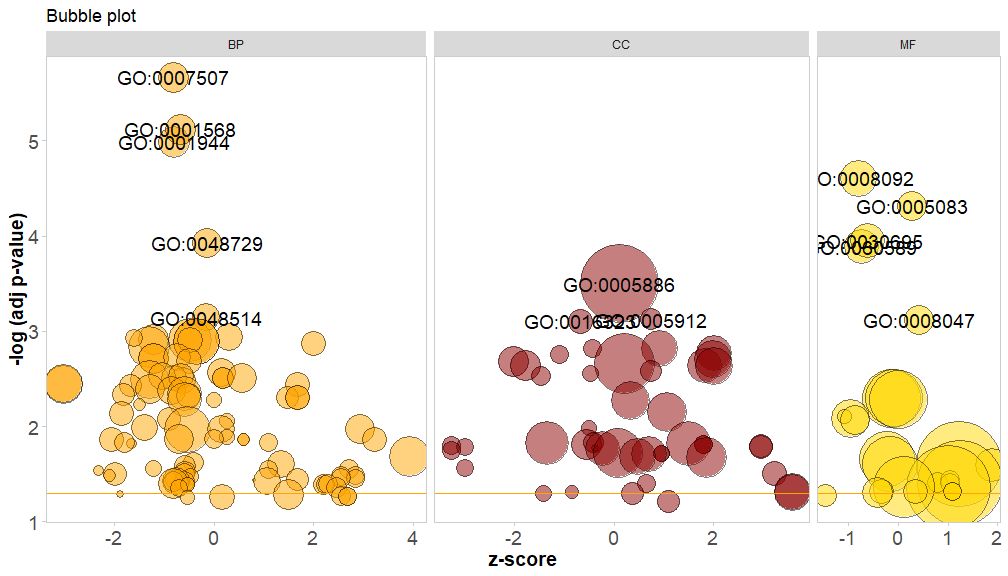
L'asse orizzontale èzscore;L'asse verticale è-log(adj p-value), simile ad un grafico a barre, più è alto, più significativo è l'arricchimento, l'area del cerchio è correlata al numero di geni nel percorso corrispondente (;circ$count ); il colore corrisponde alla categoria corrispondente al percorso, il verde è il processo biologico, il rosso è la componente cellulare e il blu è la funzione molecolare.Può essere inserito da?GOBubble Consulta la pagina di aiuto della funzione GOBubble per modificare tutti i parametri dell'immagine. Per impostazione predefinita, ogni cerchio è contrassegnato con un GO ID corrispondente e sulla destra viene visualizzata anche una tabella che mostra la relazione corrispondente tra GO ID e termine GO.I parametri possono essere impostati datable.legendperFALSE per nasconderlo. Se si desidera visualizzare la descrizione del percorso, impostare l'ID del parametro su FALSE.Tuttavia, a causa dello spazio limitato e dei cerchi sovrapposti, non tutti i cerchi sono contrassegnati, solo quelli-log(adj p-value) > 3(il valore predefinito è 5).
- # 生成泡泡图,并展示-log(adj p-value) > 3 的通路的GO ID
- GOBubble(circ, labels = 3)
Se desideri aggiungere un titolo al grafico a bolle o specificare il colore del cerchio e visualizzare i percorsi di ciascuna categoria separatamente e modificare la soglia GO ID visualizzata, puoi aggiungere i seguenti parametri:
GOBubble(circ, title = 'Bubble plot', colour = c('orange', 'darkred', 'gold'), display = 'multiple', labels = 3)
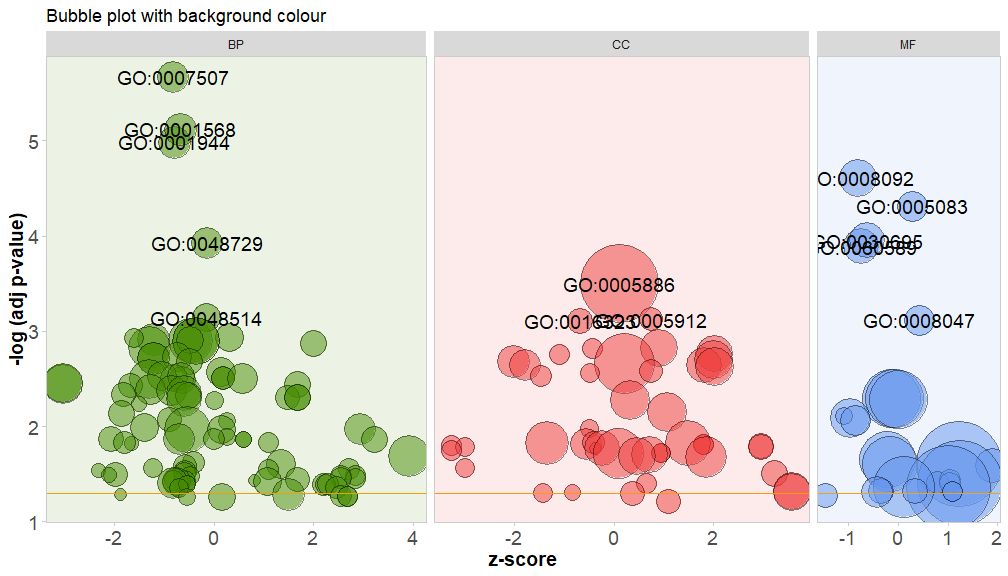
Colora lo sfondo della classe del canale impostando il parametro bg.col su TRUE.
GOBubble(circ, title = 'Bubble plot with background colour', display = 'multiple', bg.col = T, labels = 3)
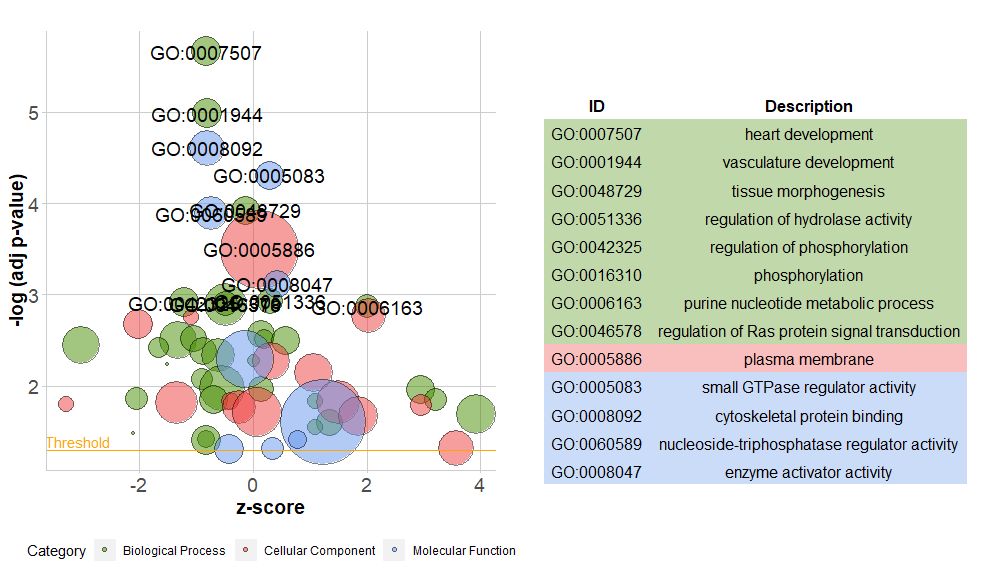
La nuova versione del pacchetto contiene una nuova funzionereduce_overlap , questa funzione può ridurre il numero di elementi ridondanti, ovvero può eliminare tutti i percorsi la cui sovrapposizione genetica è maggiore o uguale alla soglia impostata e mantenere solo un percorso di ciascun gruppo come rappresentativo, indipendentemente dalla visualizzazione di tutti percorsi in GO. Riducendo il numero di termini ridondanti, la leggibilità dei grafici (come quelli a bolle) risulta notevolmente migliorata.
- # reduce_overlap,参数设置为0.75
- reduced_circ <- reduce_overlap(circ, overlap = 0.75)
-
- GOBubble(reduced_circ, labels = 2.8)
Anche se un grafico che mostra tutte le informazioni può aiutarci a scoprire quali percorsi sono più significativi, la realtà dipende comunque dalle ipotesi e dalle idee che vuoi confermare con i dati, e i percorsi più importanti potrebbero non essere necessariamente quelli che ti interessano. Pertanto, selezionando manualmente un prezioso insieme di percorsi (EC$process ), abbiamo bisogno di un diagramma che ci mostri informazioni più dettagliate su questo specifico insieme di percorsi.Ma dalla presentazione di questi dati nasce un problema: a volte sono di difficile interpretazionezscore Informazioni fornite.Dopotutto, questo metodo di calcolo non è universale. Come mostrato sopra, è semplicemente il numero di geni sovraregolati meno il numero di geni sottoregolati diviso per la radice quadrata del numero di geni in ciascun percorso.GOCircleAnche il grafico risultante sottolinea questo fatto.
Il cerchio esterno del diagramma circolare mostra il valore logFC dei geni di ciascun percorso come punti sparsi. I cerchi rossi indicano una sovraregolazione e il blu indica una sottoregolazione.È possibile utilizzare i parametrilfc.col Cambia colore. Ciò spiega anche perché in alcuni casi percorsi molto importanti hanno zscore prossimi allo zero. Un punteggio z pari a zero non significa che il canale non è importante. Mostra semplicemente che lo zscore è una misura approssimativa, perché ovviamente anche lo zscore non tiene conto del livello funzionale e della dipendenza dall’attivazione dei singoli geni nei processi biologici.
GOCircle(circ)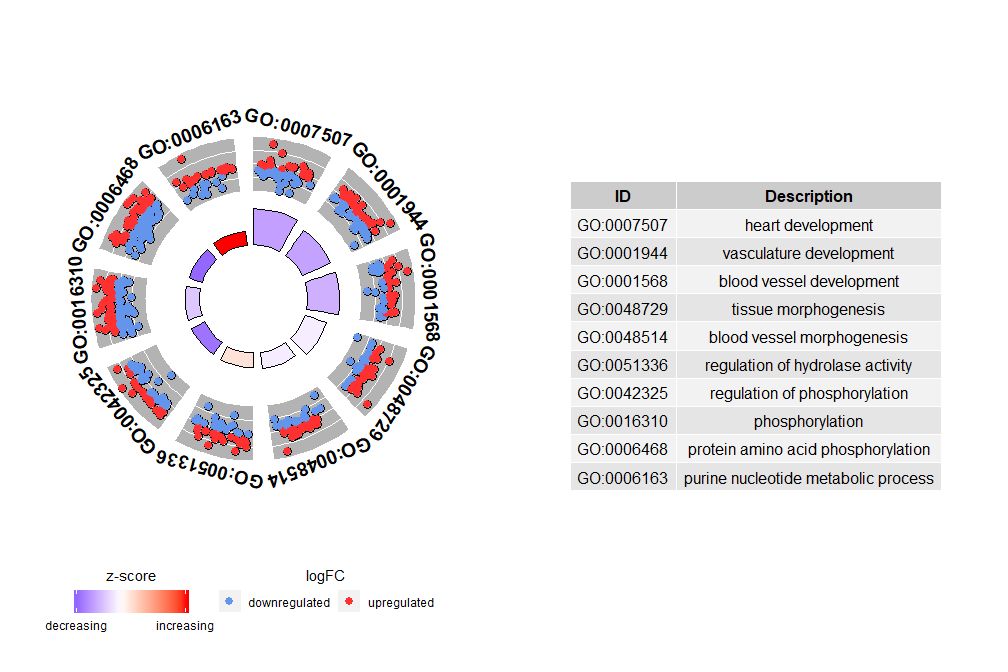
nsub I parametri possono essere numeri impostati o vettori di caratteri. Se è un vettore di caratteri, contiene il GO ID o il percorso da visualizzare;
- # 生成特定通路的圈图
- IDs <- c('GO:0007507', 'GO:0001568', 'GO:0001944', 'GO:0048729', 'GO:0048514', 'GO:0005886', 'GO:0008092', 'GO:0008047')
- GOCircle(circ, nsub = IDs)
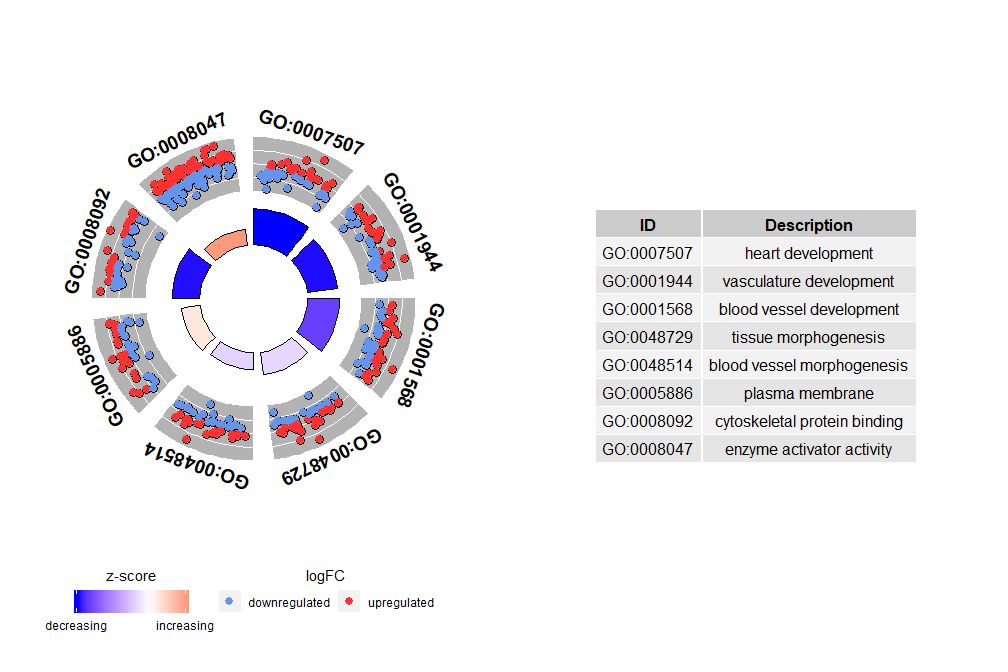
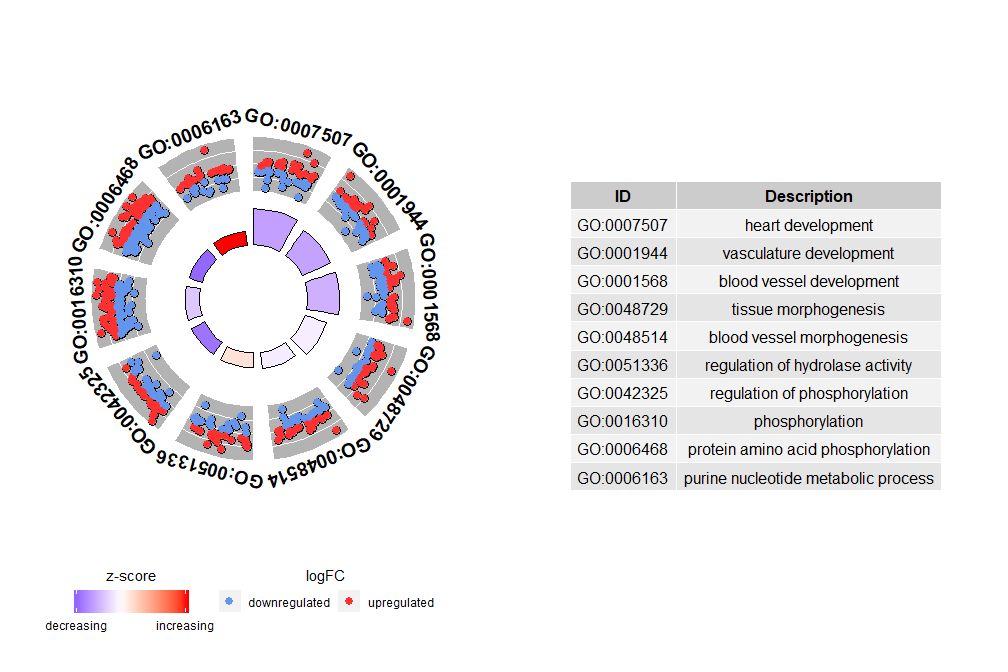
Se nsub è un vettore numerico, il numero definisce il numero da visualizzare. Inizia dalla prima riga del dataframe di input. Questa visualizzazione funziona solo con dati più piccoli. Il numero massimo di canali per impostazione predefinita è 12. Sebbene il numero di canali venga ridotto, la quantità di informazioni visualizzate aumenta.
- # 圈图展示数据前十个通路
- GOCircle(circ, nsub = 10)
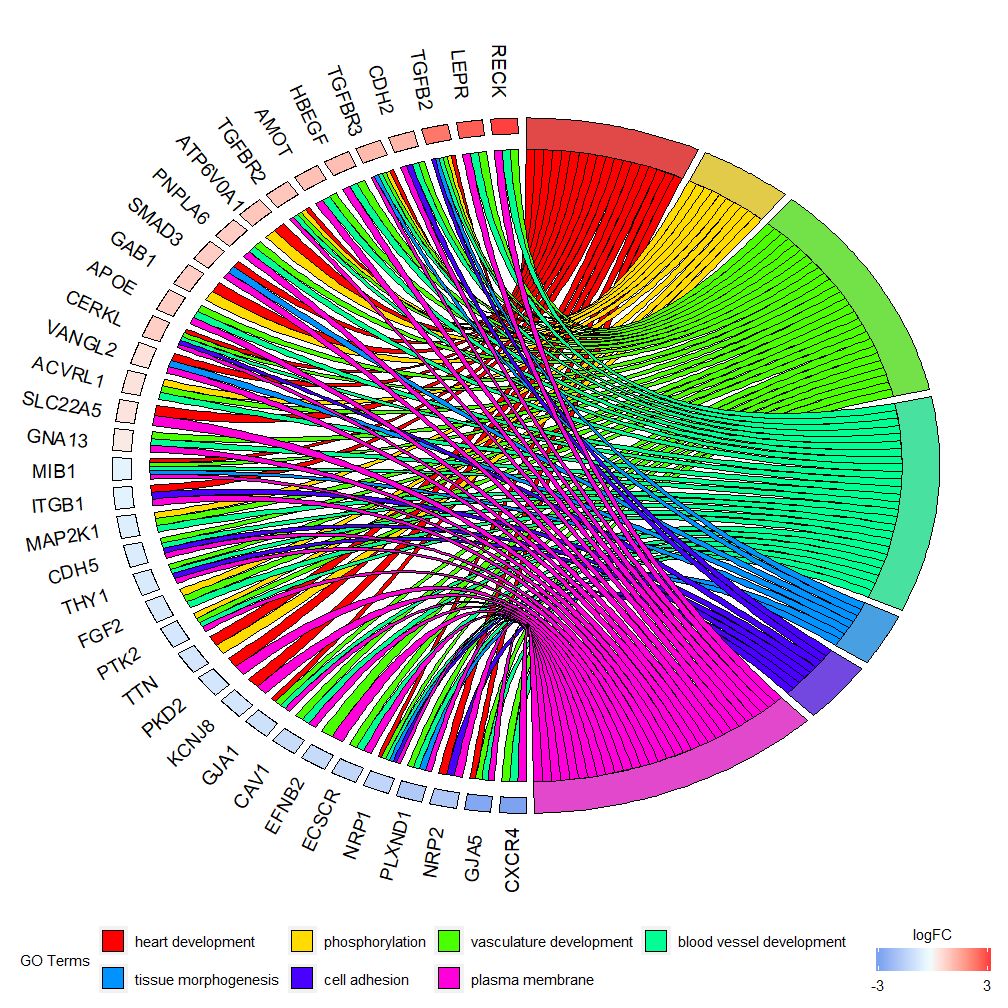
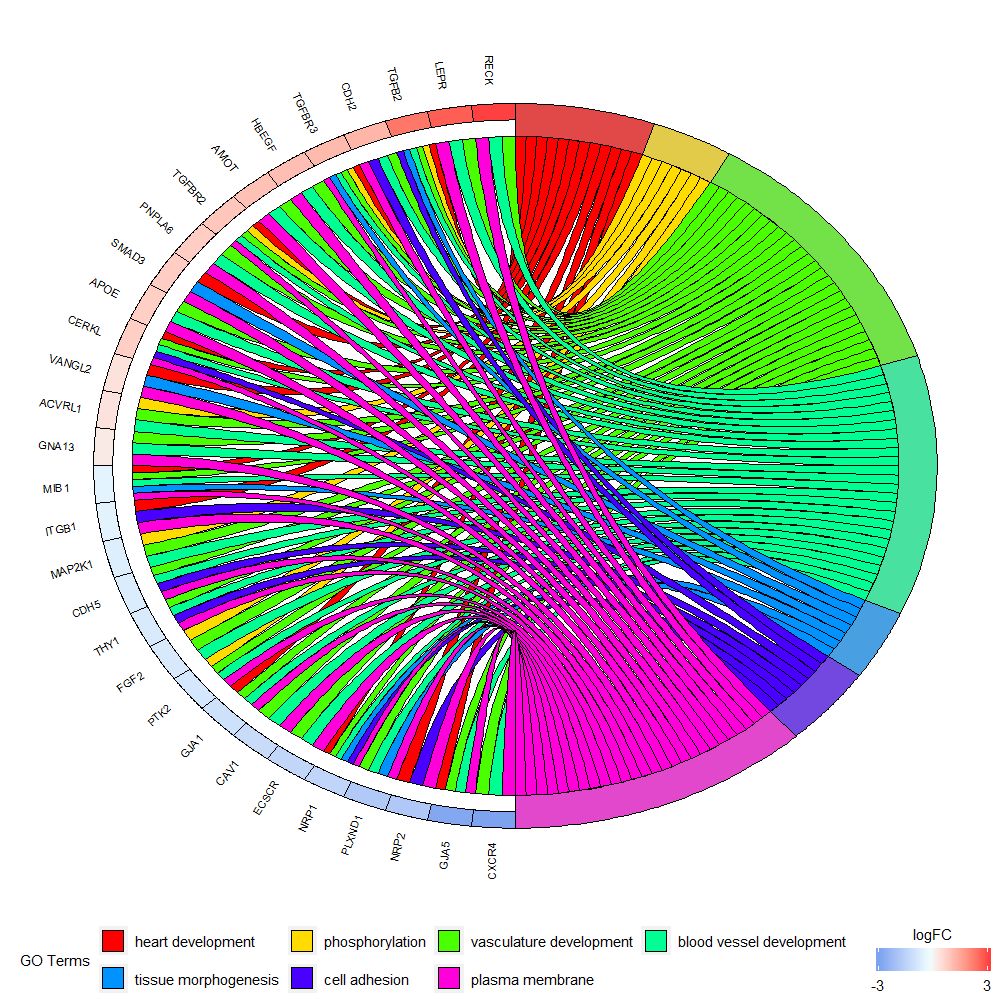
GOChord può visualizzare la relazione tra geni e percorsi selezionati e il logFC dei geni.Per prima cosa devi inserire una matrice, che puoi costruire tu stesso0-1Matrix, puoi anche utilizzare le funzionichord_dat Costruire. Questa funzione ha tre parametri: dati, geni e processo, di cui gli ultimi due parametri devono avere almeno un parametro.Quindi la funzionecircle_datCombina i dati delle espressioni con i risultati delle analisi funzionali.
I grafici a barre e a bolle possono darti una prima impressione dei dati. Ora puoi selezionare alcuni geni e percorsi che riteniamo preziosi. Sebbene GOCircle aggiunga un livello per visualizzare il valore di espressione dei geni nei percorsi, manca una singola informazione sulle relazioni tra geni e vie multiple. Non è facile capire se determinati geni sono collegati a più processi. GOChord compensa le carenze di GOCircle. Le righe dei dati generati sono geni e le colonne sono percorsi. "0" significa che il gene non è assegnato al percorso e "1" è il contrario.
- # 找到感兴趣的的基因,这里我们以EC$genes为例
- head(EC$genes)
-
- ## ID logFC
- ## 1 PTK2 -0.6527904
- ## 2 GNA13 0.3711599
- ## 3 LEPR 2.6539788
- ## 4 APOE 0.8698346
- ## 5 CXCR4 -2.5647537
- ## 6 RECK 3.6926860
-
- # 获得感兴趣基因的通路
- EC$process
-
- ## [1] "heart development" "phosphorylation"
- ## [3] "vasculature development" "blood vessel development"
- ## [5] "tissue morphogenesis" "cell adhesion"
- ## [7] "plasma membrane"
-
- # 使用chord_dat构建矩阵
- chord <- chord_dat(circ, EC$genes, EC$process)
- head(chord)
-
- ## heart development phosphorylation vasculature development
- ## PTK2 0 1 1
- ## GNA13 0 0 1
- ## LEPR 0 0 1
- ## APOE 0 0 1
- ## CXCR4 0 0 1
- ## RECK 0 0 1
- ## blood vessel development tissue morphogenesis cell adhesion
- ## PTK2 1 0 0
- ## GNA13 1 0 0
- ## LEPR 1 0 0
- ## APOE 1 0 0
- ## CXCR4 1 0 0
- ## RECK 1 0 0
- ## plasma membrane logFC
- ## PTK2 1 -0.6527904
- ## GNA13 1 0.3711599
- ## LEPR 1 2.6539788
- ## APOE 1 0.8698346
- ## CXCR4 1 -2.5647537
- ## RECK 1 3.6926860
Nell'esempio abbiamo passato due parametri. Se viene specificato solo il parametro genes, il risultato è un elenco di geni selezionati e tutte le costruzioni del processo con almeno un gene specificato.0-1matrice; se solo specificatoprocessparametri, il risultato è che tutti i geni generano0-1 Matrice di geni assegnati ad almeno un processo nell'elenco. Tenere presente che specificare solo i geni e i parametri di processo può comportare una matrice 0-1 molto grande, con conseguenti risultati di visualizzazione confusi.
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
-
- # Generate the matrix with a list of selected genes
- chord_genes <- chord_dat(data = circ, genes = EC$genes)
- head(chord_genes)
-
- ## heart development vasculature development blood vessel development
- ## PTK2 0 1 1
- ## GNA13 0 1 1
- ## LEPR 0 1 1
- ## APOE 0 1 1
- ## CXCR4 0 1 1
Questo grafico ha lo scopo di mostrare un sottoinsieme più piccolo di dati ad alta dimensione. I parametri che possono essere regolati sono principalmente due:gene.orderEnlfc . Il parametro dei geni può essere specificato come 'logFC', 'alfabetico', 'nessuno'. Infatti, generalmente specifichiamo il parametro dei geni come logFC; il parametro nlfc è uno dei parametri più importanti di questa funzione, perché può gestire il modo in cui ciascun gene ha 0 o più valori logFC presentati nella matrice. Pertanto dovremmo specificare i parametri per evitare errori.
Ad esempio, se hai una matrice senza valori logFC, devi impostarenlfc=0 ; Oppure eseguire l'analisi dell'espressione differenziale sui geni in più condizioni o lotti. In questo caso, ciascun gene contiene più valori logFC ed è necessario impostare il numero di colonna nlfc=logFC. Il valore predefinito è "1" perché si ritiene che nella maggior parte dei casi ci sarà un solo valore logFC per gene. Utilizza il parametro space per definire lo spazio tra i rettangoli colorati che rappresentano logFC. Il parametro gene.size specifica la dimensione del carattere del nome del gene e gene.space specifica la dimensione dello spazio tra i nomi dei geni.
- chord <- chord_dat(data = circ, genes = EC$genes, process = EC$process)
- GOChord(chord, space = 0.02, gene.order = 'logFC', gene.space = 0.25, gene.size = 5)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
Può essere impostato in base al valore logFCgene.order=‘logFC’ , i geni vengono ordinati in base ai loro valori logFC. A volte l'immagine può diventare un po' affollata e questo può essere automatizzato utilizzando il parametro limite per ridurre il numero di geni o percorsi mostrati. Limit è un vettore con due valori di cutoff (il valore predefinito è c(0,0)). Il primo valore specifica il numero minimo di percorsi a cui deve essere assegnato il gene. Il secondo valore determina il numero di geni assegnati al percorso.
- # 仅显示分配给至少三个通路的基因
- GOChord(chord, limit = c(3, 0), gene.order = 'logFC')
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
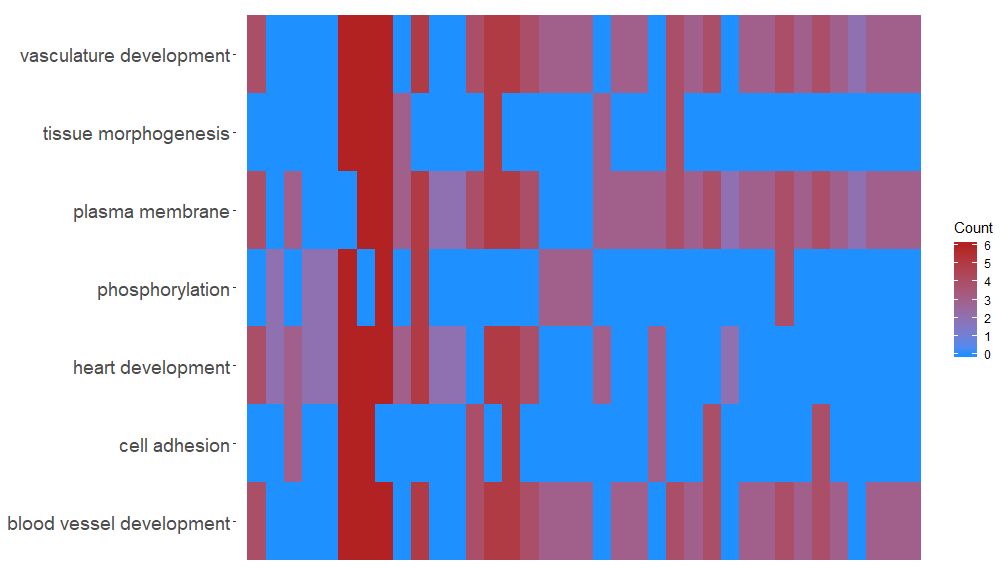
La funzione GOHeat può visualizzare la relazione tra geni e percorsi utilizzando una mappa termica, simile a GOChord. I processi biologici vengono visualizzati orizzontalmente e i geni vengono visualizzati verticalmente. Ogni colonna è divisa in piccoli rettangoli e il colore generalmente dipende dal valore logFC. Inoltre, i geni arricchiti in percorsi funzionali simili sono stati raggruppati. Sono disponibili due modalità per la selezione del colore della mappa termica, a seconda dei parametri nlfc. Se nlfc = 0, il colore è il numero di percorsi arricchiti per ciascun gene. Vedi esempi per i dettagli:
- # First, we use the chord object without logFC column to create the heatmap
- GOHeat(chord[,-8], nlfc = 0)
GOHeat(chord[,-8])Il colore corrisponde al logFC del gene nel caso nlfc = 1
GOHeat(chord, nlfc = 1, fill.col = c('red', 'yellow', 'green'))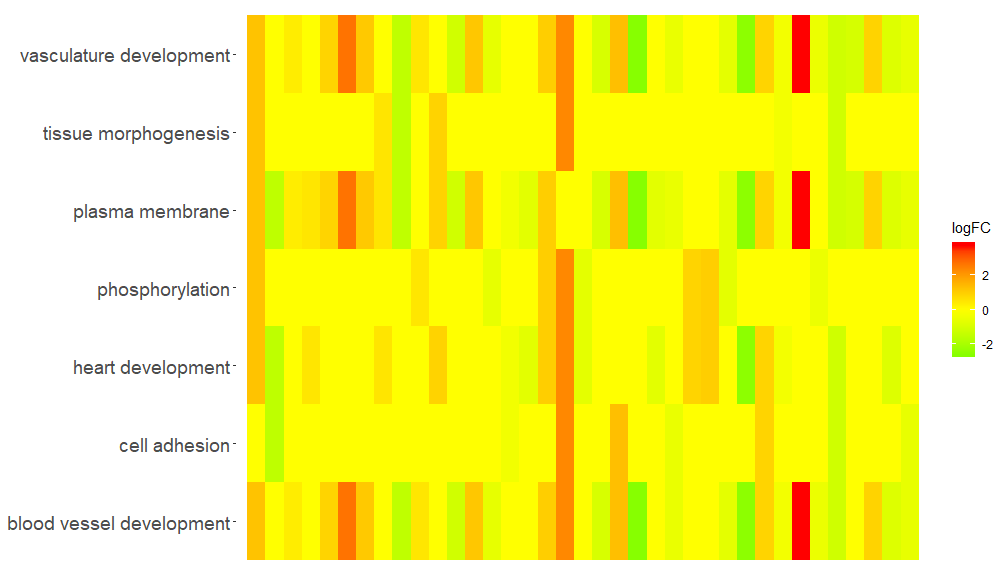
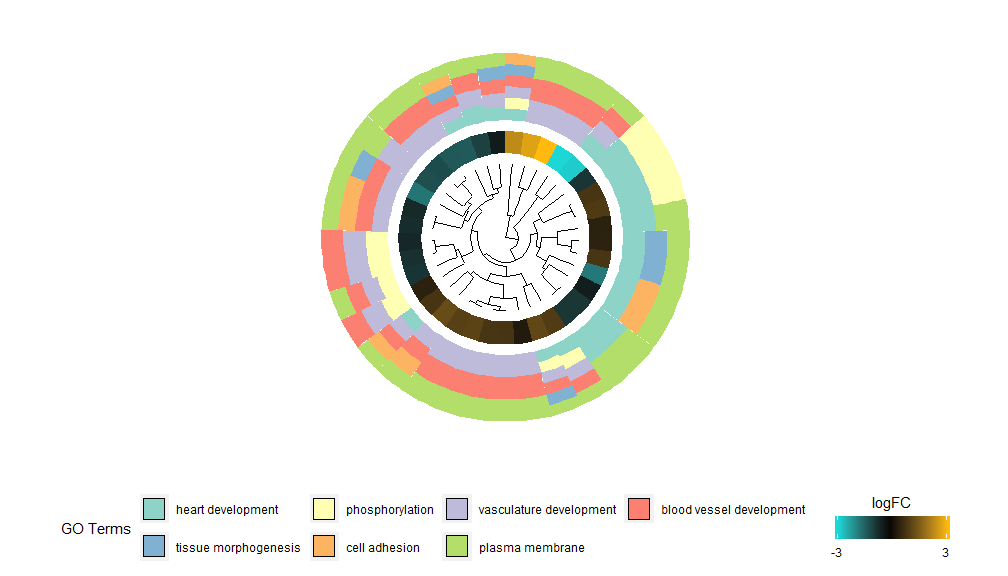
L'idea alla base della funzionalità GOCluster è visualizzare quante più informazioni possibili. Ecco un esempio:
- GOCluster(circ, EC$process, clust.by = 'logFC', term.width = 2)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
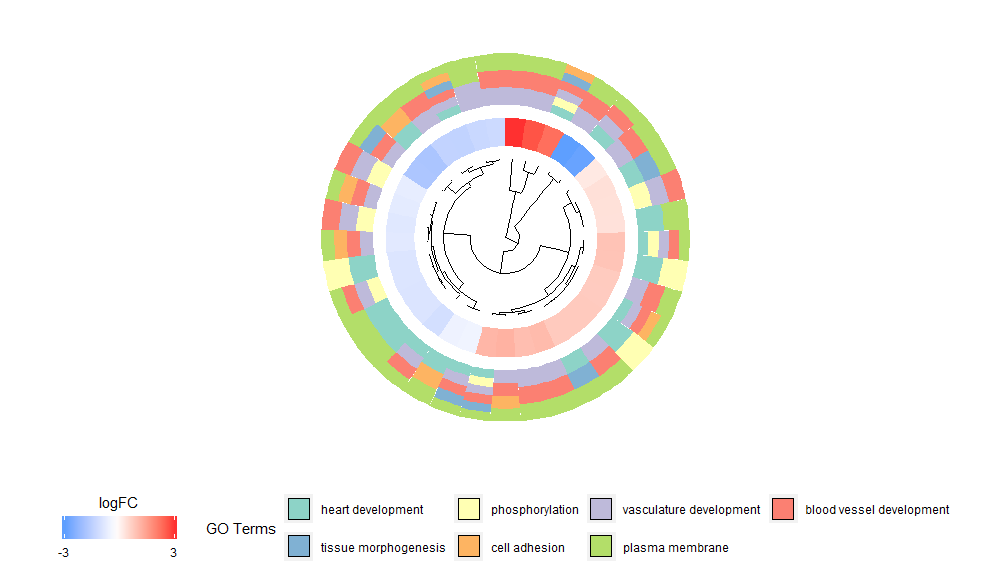
Il clustering gerarchico è un popolare metodo di analisi di clustering non supervisionato per l'espressione genica che garantisce un raggruppamento imparziale di geni insieme in base al modello di espressione, in modo che i cluster possano contenere più gruppi di geni co-regolati o funzionalmente correlati. GOCluster utilizza il filehclust Il metodo esegue il clustering gerarchico dei profili di espressione genica. Se desideri modificare la metrica della distanza o l'algoritmo di clustering, utilizza rispettivamente i parametri metric e clust. Il dendrogramma risultante può essere convertito con l'aiuto di ggdendro e visualizzato con ggplot2. Scegli un layout circolare poiché non è solo efficace ma anche visivamente accattivante. Il primo cerchio accanto al dendrogramma rappresenta il logFC del gene, che in realtà è la foglia dell'albero del clustering. Se sei interessato a contrasti multipli puoi modificare il parametro nlfc, di default è impostato su "1" quindi viene disegnato un solo anello. I valori logFC sono codificati a colori utilizzando una scala di colori definibile dall'utente (lfc.col); il cerchio successivo rappresenta il percorso assegnato al gene. Per avere un bell'aspetto, il numero di canali è stato ridotto e il colore dei canali può essere modificato utilizzando il parametro term.col.ancora disponibile?GOCluster per vedere come modificare i parametri. Il parametro più importante di questa funzione è cluster.by, che può essere specificato per raggruppare in base a modelli di espressione genetica ("logFC", come mostrato sopra) o categorie funzionali ("termini").
- GOCluster(circ, EC$process, clust.by = 'term', lfc.col = c('darkgoldenrod1', 'black', 'cyan1'))
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
I diagrammi di Venn possono essere utilizzati per rilevare le relazioni tra vari elenchi di geni espressi in modo differenziale o per esplorare l'intersezione di geni di percorsi multipli nelle analisi funzionali. I diagrammi di Venn non mostrano solo il numero di geni sovrapposti, ma anche informazioni sul modello di espressione del gene (solitamente sovraregolato, spesso sottoregolato o controregolato). Attualmente vengono utilizzati fino a tre set di dati come input. I dati di input contengono almeno due colonne: una per i nomi dei geni e una per i valori logFC.
- l1 <- subset(circ, term == 'heart development', c(genes,logFC))
- l2 <- subset(circ, term == 'plasma membrane', c(genes,logFC))
- l3 <- subset(circ, term == 'tissue morphogenesis', c(genes,logFC))
- GOVenn(l1,l2,l3, label = c('heart development', 'plasma membrane', 'tissue morphogenesis'))
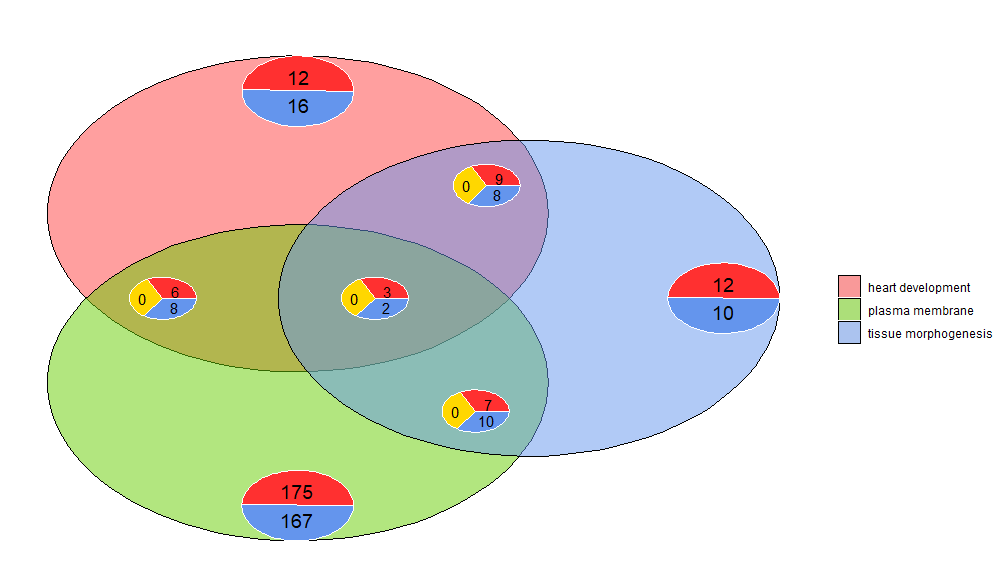
Ad esempio, lo sviluppo cardiaco e la morfogenesi dei tessuti hanno 22 geni, 12 sono sovraregolati e 10 sottoregolati. Una cosa importante da notare è che i grafici a torta non visualizzano informazioni ridondanti. Pertanto, se si confrontano tre set di dati, i geni comuni a tutti i set di dati (il grafico a torta centrale) non sono inclusi negli altri grafici a torta. Questo strumento è disponibile su ShinyApp https://wwalter.shinyapps.io/Venn/, lo strumento web è più interattivo, il cerchio ha un'area proporzionale al numero di geni nel set di dati e il cursore può essere utilizzato per spostare il piccolo grafico a torta e dispone di GOVenn Features tutte le opzioni per modificare il layout della trama e anche per scaricare immagini ed elenchi di geni.
Home page del software: https://wencke.github.io/

Si dedica alla ricerca tecnologica da più di 30 anni ed è esperto in vari linguaggi come Java, Linux, Javascript, php, css, ecc. Ha dato numerosi contributi nel campo dell'open source stazione di documentazione per gli sviluppatori per condividere alcuni problemi nello sviluppo della tecnologia per riferimento futuro

Posta[email protected]