
informasi kontak saya
Surat[email protected]
2024-07-12
한어Русский языкEnglishFrançaisIndonesianSanskrit日本語DeutschPortuguêsΕλληνικάespañolItalianoSuomalainenLatina
Paket GOPlot digunakan untuk visualisasi data biologis. Sebaliknya, paket ini mengintegrasikan dan memvisualisasikan data ekspresi dengan hasil analisis fungsional.Tetapi berhati-hatilahPaket ini tidak dapat digunakan untuk melakukan analisis ini, hanya untuk memvisualisasikan hasilnya. . Di semua bidang ilmu pengetahuan, sulit untuk menggambarkan sesuatu secara realistis karena keterbatasan ruang dan kesederhanaan yang diperlukan untuk mendapatkan hasil, sehingga informasi perlu divisualisasikan dan gambar digunakan untuk menyampaikan informasi. Grafik yang dirancang dengan baik memberikan lebih banyak informasi dalam ruang yang lebih sedikit. Ide dari paket ini adalah untuk memungkinkan pengguna dengan cepat memeriksa data dalam jumlah besar, mengungkapkan tren dalam data dan menemukan pola dan korelasi dalam data.
Visualisasi data dapat membantu kita menemukan jawaban atas pertanyaan biologis, menilai hipotesis tertentu, dan bahkan menemukan sudut pandang berbeda untuk menyelidiki masalah yang berbeda. Dan fungsi plot dari paket ini dikembangkan berdasarkan struktur hierarki data, dimulai dengan keseluruhan data dan diakhiri dengan subset gen yang dipilih dan jalur yang sesuai.
Mari kita jelaskan secara konkrit dengan sebuah contoh.
Kami menyebut data yang datang dengan GOplot, yang berasal dari GEOGSE47067, berisi informasi transkriptom sel endotel dari dua jaringan (otak dan jantung). Untuk informasi lebih lanjut, lihat makalah oleh Nolan et al.Data dinormalisasi dan gen yang diekspresikan berbeda ditemukan., lalu gunakan alat anotasi fungsi DAVID (data anotasi DAVID diperbarui secara perlahan dan tidak disarankan saat ini. Disarankan untuk menggunakanGo East, alat analisis pengayaan GO online terbaikDanWebsite yang dapat melakukan analisis pengayaan hanya dalam satu langkah ini telah dikutip lebih dari 350 kali oleh CNS dan lainnya sebelum dipublikasikan.Lakukan analisis pengayaan,Kuasai GSEA dalam satu artikel, tutorial super detail) Anotasi gen dari gen yang diekspresikan secara berbeda (adjusted p-value < 0.05 ) dan analisis pengayaan fungsional. Kumpulan data ini berisi lima kategori data berikut:
| nama | menggambarkan | Ukuran kumpulan data |
|---|---|---|
| EC$eset | Ekspresi gen yang dinormalisasi dalam sel endotel otak dan jantung (3 ulangan) | 20644x7 tahun |
| Daftar EC$generik | Gen yang diekspresikan secara berbeda (nilai p yang disesuaikan <0,05) | 2039x7 tahun |
| EC$david | Hasil analisis pengayaan fungsional gen diferensial menggunakan DAVID | Ukuran 174x5 |
| gen EC$ | Gen dan logFC | ukuran 37x2 |
| Proses EC$ | Vektor fitur terpilih untuk proses biologis yang diperkaya | 7 |
Kami ingin melihat jalur gen yang diekspresikan secara berbeda yang diperkaya dengan GO, namun sebelum mulai menggambar, kami perlu menyediakan data yang memenuhi persyaratan format.Secara umum, data yang diperlukan untuk menggambar grafik disediakan sendiri, tapiAda fungsi dalam paket inicircle_datDapat membantu kami menangani format data。circle_datIni dapat menggabungkan hasil analisis pengayaan fungsional gen yang dipilih dan nilai logFC-nya, terutama untuk gen yang diekspresikan secara berbeda.circle_dat Penggunaannya sangat sederhana, cukup membaca dua data. Data pertama berisi hasil analisis pengayaan fungsional, dengan minimal empat kolom (kategori analisis pengayaan fungsional, jalur, gen, nilai p yang disesuaikan).Data kedua adalah gen yang dipilih dan logFC-nya, data ini bisa menjadi sumbernyalimmaHasil analisis statistik (Catatan dari Biografi: Pastikan untuk memperhatikan dua fileBagaimana gen diberi namaBersikaplah konsisten, seperti semua orangGene symbol ). Mari kita lihat format data yang disebutkan di atas dengan contoh.
- #安装已发布的稳定版本
- #install.packages('GOplot')
- #安装github上的开发版本
- #install_github('wencke/wencke.github.io')
- #载入包
- library(GOplot)
- #读入包内自带的数据
- data(EC)
- #查看功能富集分析结果的数据格式
- head(EC$david)
-
- ## Category ID Term
- ## 1 BP GO:0007507 heart development
- ## 2 BP GO:0001944 vasculature development
- ## 3 BP GO:0001568 blood vessel development
- ## 4 BP GO:0048729 tissue morphogenesis
- ## 5 BP GO:0048514 blood vessel morphogenesis
- ## 6 BP GO:0051336 regulation of hydrolase activity
- ## Genes
- ## 1 DLC1, NRP2, NRP1, EDN1, PDLIM3, GJA1, TTN, GJA5, ZIC3, TGFB2, CERKL, GATA6, COL4A3BP, GAB1, SEMA3C, MKL2, SLC22A5, MB, PTPRJ, RXRA, VANGL2, MYH6, TNNT2, HHEX, MURC, MIB1, FOXC2, FOXC1, ADAM19, MYL2, TCAP, EGLN1, SOX9, ITGB1, CHD7, HEXIM1, PKD2, NFATC4, PCSK5, ACTC1, TGFBR2, NF1, HSPG2, SMAD3, TBX1, TNNI3, CSRP3, FOXP1, KCNJ8, PLN, TSC2, ATP6V0A1, TGFBR3, HDAC9
- ## 2 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, EFNB2, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 3 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 4 DLC1, ENAH, NRP1, PGF, ZIC2, TGFB2, CD44, ILK, SEMA3C, RET, AR, RXRA, VANGL2, LEF1, TNNT2, HHEX, MIB1, NCOA3, FOXC2, FOXC1, TGFB1I1, WNT5A, COBL, BBS4, FGFR3, TNC, BMPR2, CTNND1, EGLN1, NR3C1, SOX9, TCF7L1, IGF1R, FOXQ1, MACF1, HOXA5, BCL2, PLXND1, CAR2, ACTC1, TBX4, SMAD3, FZD3, SHANK3, FZD6, HOXB4, FREM2, TSC2, ZIC5, TGFBR3, APAF1
- ## 5 GNA13, CAV1, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, CDH2, MEIS1, WT1, TGFB2, WARS, PTK2, CERKL, APOE, CXCR4, ANG, SEMA3C, PLCD1, NOS2, MKL2, PLXND1, FIGF, FGF2, PTPRJ, TGFBR2, TBX4, NF1, TBX1, TNNI3, PNPLA6, VASH1, THY1, NUS1, MEOX2, ECSCR, AMOT, HBEGF, FOXC2, FOXC1, HDAC7
- ## 6 CAV1, XIAP, AGFG1, ADORA2A, TNNC1, TBC1D9, LEPR, ABHD5, EDN1, ASAP2, ASAP3, SMAP1, TBC1D12, ANG, TBC1D14, MTCH1, TBC1D13, TBC1D4, TBC1D30, DHCR24, HIP1, VAV3, NOS1, NF1, MYH6, RICTOR, TBC1D22A, THY1, PLCE1, RNF7, NDEL1, CHML, IFT57, ACAP2, TSC2, ERN1, APAF1, ARAP3, ARAP2, ARAP1, HTR2A, F2R
- ## adj_pval
- ## 1 0.000002170
- ## 2 0.000010400
- ## 3 0.000007620
- ## 4 0.000119000
- ## 5 0.000720000
- ## 6 0.001171166
-
- #查看基因的数据格式
- head(EC$genelist)
-
- ## ID logFC AveExpr t P.Value adj.P.Val B
- ## 1 Slco1a4 6.645388 1.2168670 88.65515 1.32e-18 2.73e-14 29.02715
- ## 2 Slc19a3 6.281525 1.1600468 69.95094 2.41e-17 2.49e-13 27.62917
- ## 3 Ddc 4.483338 0.8365231 65.57836 5.31e-17 3.65e-13 27.18476
- ## 4 Slco1c1 6.469384 1.3558865 59.87613 1.62e-16 8.34e-13 26.51242
- ## 5 Sema3c 5.515630 2.3252117 58.53141 2.14e-16 8.81e-13 26.33626
- ## 6 Slc38a3 4.761755 0.9218670 54.11559 5.58e-16 1.76e-12 25.70308
Setelah memahami kedua format data input tersebut, Anda dapat menggunakannyacirlce_datberfungsi untuk menghasilkan data gambar.
- # 生成画图所需的数据格式
- circ <- circle_dat(EC$david, EC$genelist)
-
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
circObjek tersebut memiliki delapan kolom data yaitu
kategori: BP (Proses Biologis), CC (Komponen Seluler) atau MF (Fungsi Molekuler)
ID: GO id (kolom opsional, jika ingin menggunakan alat analisis fungsional yang tidak berbasis GO id, kolom ID tidak dapat dipilih; ID di sini juga dapat berupa KEGG ID)
istilah: jalur GO
count: jumlah gen di setiap jalur
gen: nama gen - logFC: nilai logFC setiap gen
adj_pval: nilai p yang disesuaikan, jalur dengan adj_pval<0,05 dianggap diperkaya secara signifikan
zscore: zscore tidak mengacu pada metode normalisasi statistik, tetapi merupakan nilai yang mudah dihitung untuk memperkirakan apakah suatu proses biologis (/fungsi molekul/komponen seluler) lebih cenderung menurun (nilai negatif) atau meningkat (nilai positif).Cara penghitungannya adalah jumlah gen yang terregulasi ke atas dikurangi jumlah gen yang terregulasi ke bawah dibagi akar kuadrat jumlah gen di setiap jalur.
Saat pertama kali melihat data, kami ingin menampilkan sebanyak mungkin jalur dari grafik, dan kami juga ingin menemukan jalur yang berharga, jadi kami memerlukan beberapa parameter untuk mengevaluasi kepentingannya. Diagram batang sering kali digunakan untuk mendeskripsikan data sampel, sehingga kita dapat menggunakan fungsi GOBar untuk membuat diagram batang yang terlihat bagus dengan cepat.
Pertama, diagram batang sederhana dibuat secara langsungGO Terms, menurut merekazscoreUrutkan batangnya; sumbu vertikalnya adalah-log(adj p-value);Warna mewakilizscore, biru menunjukkanz-scoreadalah nilai negatif, ekspresi gen pada jalur terkait cenderung menurun, ditandai dengan warna merahz-score Jika bernilai positif, ekspresi gen pada jalur terkait kemungkinan besar akan meningkat. Jika diinginkan, urutan dapat diubah dengan mengatur parameter order.by.zscore ke FALSE, dalam hal ini batang diurutkan berdasarkan signifikansinya.
- # 生成简单的条形图
- GOBar(subset(circ, category == 'BP'))
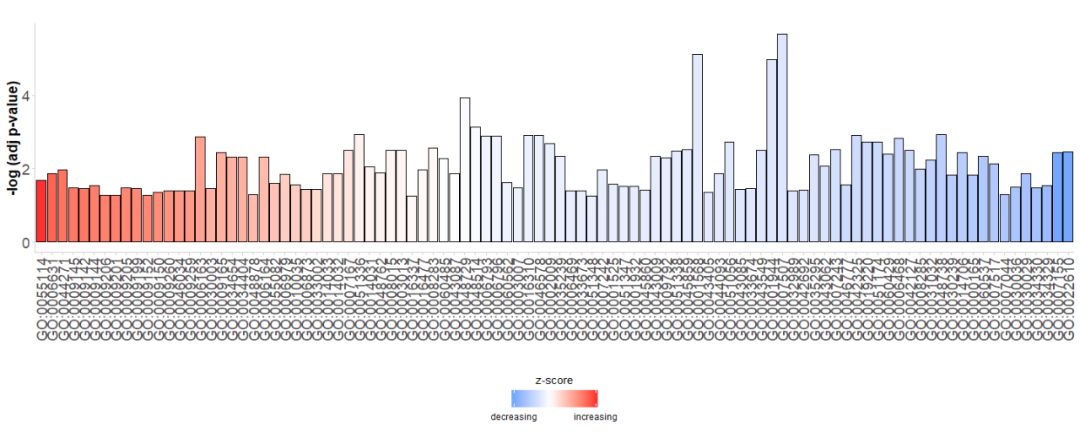
#GOBar(subset(circ, category == 'BP',order.by.zscore=FALSE))Selain itu, ubah parameter tampilan untuk menggambar diagram batang sesuai dengan kategori saluran.
- #根据通路的类别来绘制条形图
- GOBar(circ, display = 'multiple')
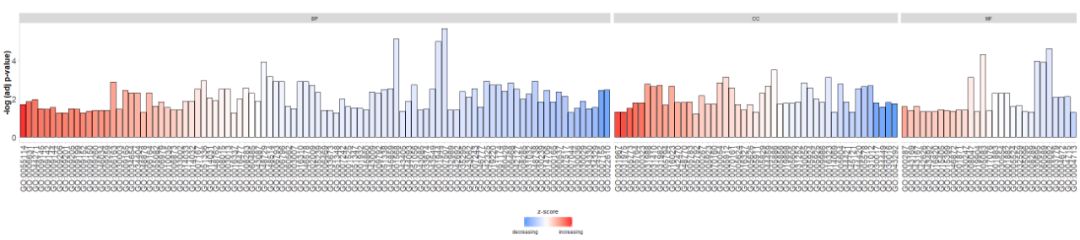
Tambahkan judul dan gunakan parameterzsc.colMengubahzscorewarna.
- # Facet the barplot, add a title and change the colour scale for the z-score
- GOBar(circ, display = 'multiple', title = 'Z-score coloured barplot', zsc.col = c('yellow', 'black', 'cyan'))
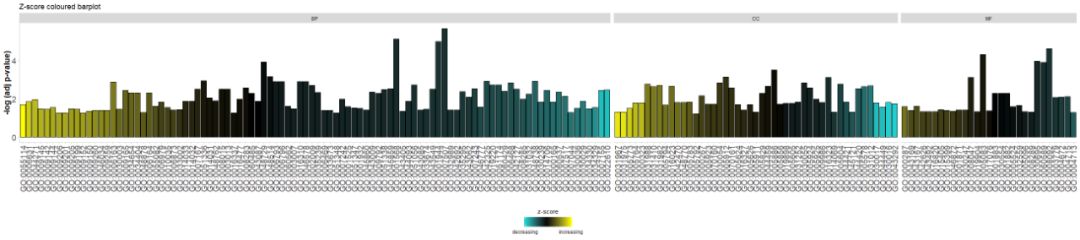
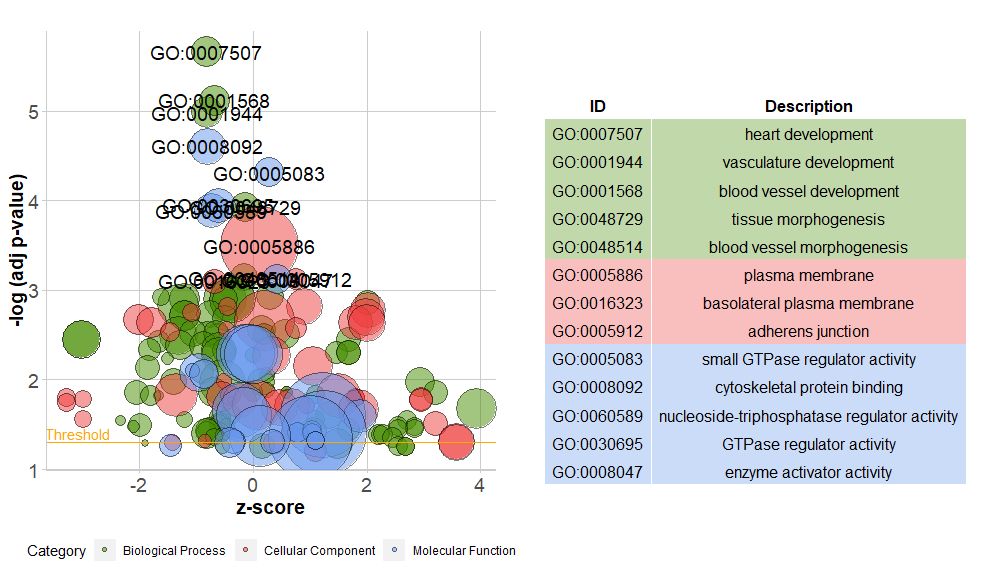
Diagram batang sangat umum dan mudah dipahami, namun kita dapat menggunakan diagram gelembung untuk menampilkan lebih banyak informasi tentang data.
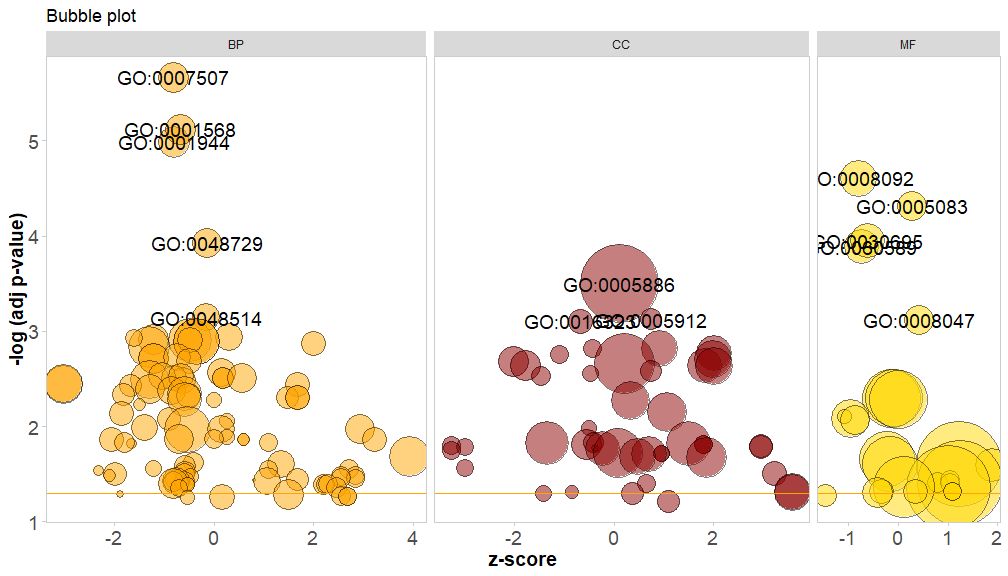
Sumbu horizontalnya adalahzscore;Sumbu vertikal adalah-log(adj p-value), mirip dengan diagram batang, semakin tinggi, semakin signifikan pengayaannya; luas lingkaran berhubungan dengan jumlah gen pada jalur yang bersangkutan (circ$count ); warna sesuai dengan kategori yang sesuai dengan jalurnya, hijau adalah proses biologis, merah adalah komponen seluler, dan biru adalah fungsi molekuler.Dapat dimasuki oleh?GOBubble Lihat halaman bantuan fungsi GOBubble untuk mengubah semua parameter gambar. Secara default, setiap lingkaran ditandai dengan GO ID yang sesuai, dan tabel yang menunjukkan hubungan yang sesuai antara GO ID dan istilah GO juga ditampilkan di sebelah kanan.Parameter dapat diatur dengantable.legenduntukFALSE untuk menyembunyikannya. Jika Anda ingin menampilkan deskripsi jalur, atur ID parameter ke FALSE.Namun karena keterbatasan ruang dan lingkaran yang tumpang tindih, tidak semua lingkaran diberi tanda, hanya lingkaran saja-log(adj p-value) > 3(standarnya adalah 5).
- # 生成泡泡图,并展示-log(adj p-value) > 3 的通路的GO ID
- GOBubble(circ, labels = 3)
Jika Anda ingin menambahkan judul pada diagram gelembung, atau menentukan warna lingkaran dan menampilkan jalur masing-masing kategori secara terpisah dan mengubah ambang batas GO ID yang ditampilkan, Anda dapat menambahkan parameter berikut:
GOBubble(circ, title = 'Bubble plot', colour = c('orange', 'darkred', 'gold'), display = 'multiple', labels = 3)
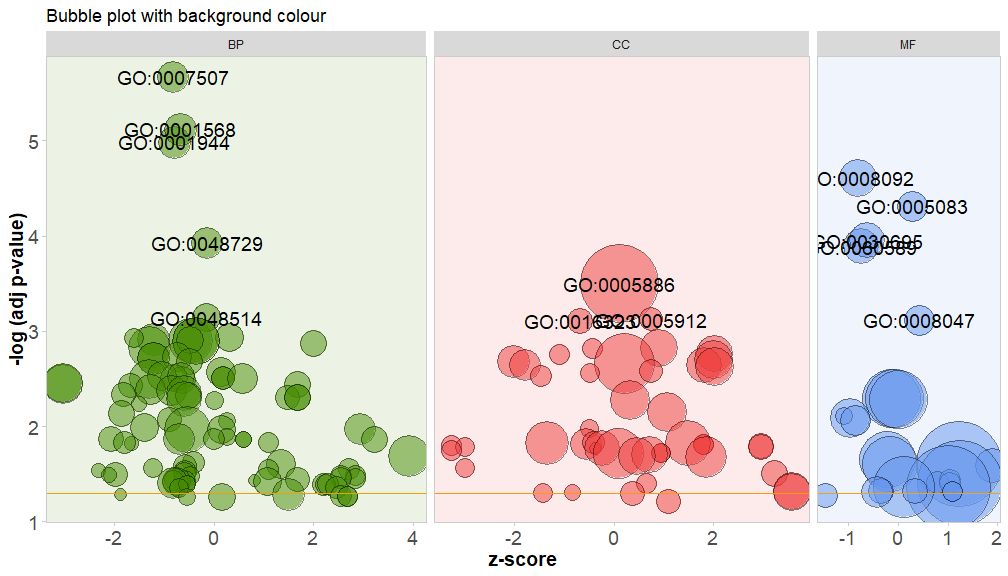
Warnai latar belakang kelas saluran dengan mengatur parameter bg.col ke TRUE.
GOBubble(circ, title = 'Bubble plot with background colour', display = 'multiple', bg.col = T, labels = 3)
Versi baru paket berisi fungsi barureduce_overlap , fungsi ini dapat mengurangi jumlah item yang berlebihan, yaitu, dapat menghapus semua jalur yang tumpang tindih gennya lebih besar dari atau sama dengan ambang batas yang ditetapkan, dan hanya mempertahankan satu jalur dari setiap grup sebagai perwakilan, terlepas dari tampilan semuanya. jalur di GO. Dengan mengurangi jumlah istilah yang berlebihan, keterbacaan plot (seperti plot gelembung) meningkat secara signifikan.
- # reduce_overlap,参数设置为0.75
- reduced_circ <- reduce_overlap(circ, overlap = 0.75)
-
- GOBubble(reduced_circ, labels = 2.8)
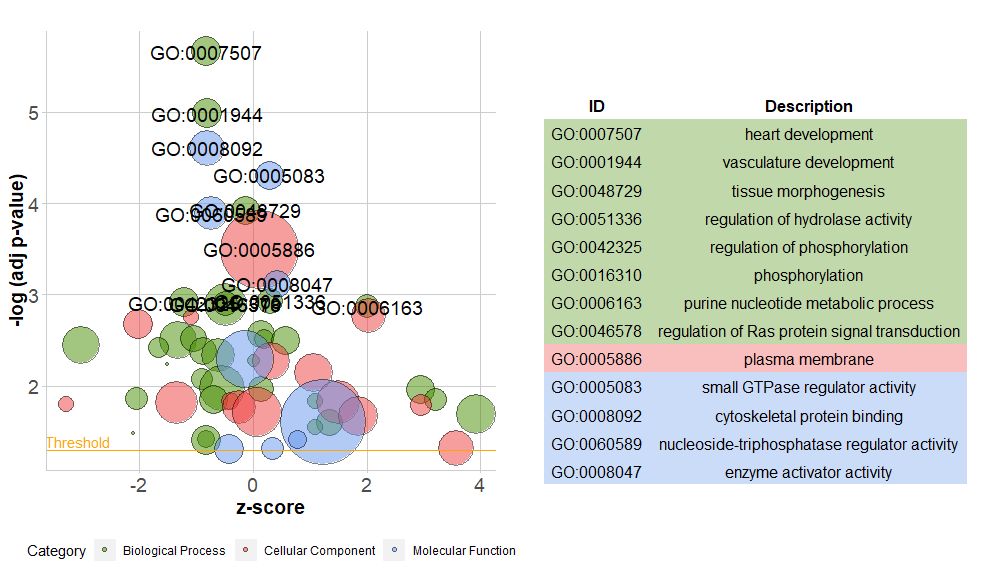
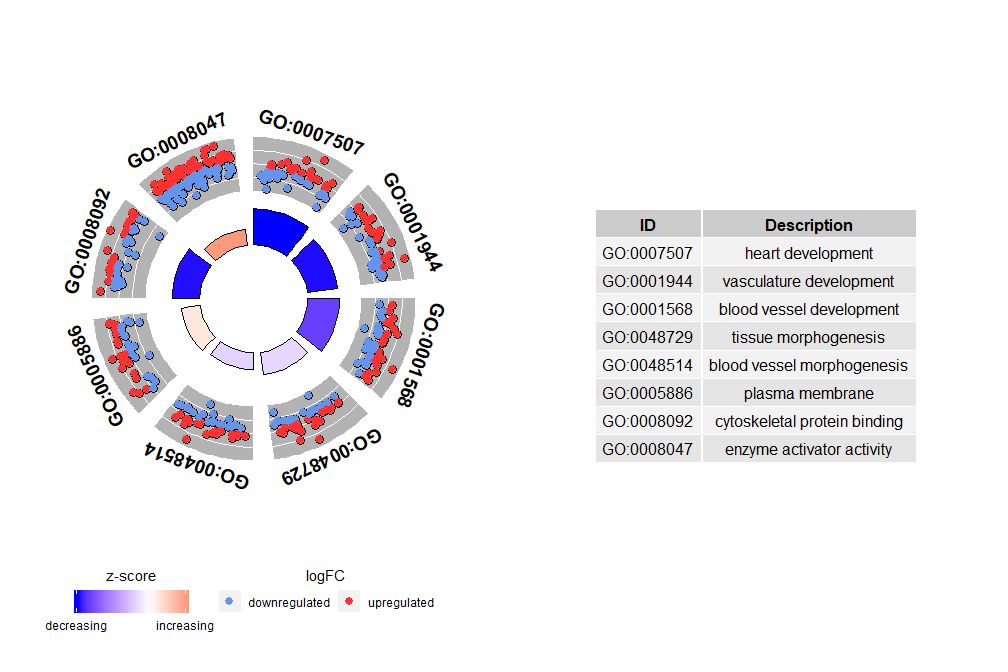
Meskipun grafik yang menunjukkan semua informasi dapat membantu kita menemukan jalur mana yang paling bermakna, kenyataannya tetap bergantung pada hipotesis dan gagasan yang ingin Anda konfirmasikan dengan data, dan jalur yang paling penting belum tentu merupakan jalur yang Anda minati. Oleh karena itu, dalam memilih secara manual serangkaian jalur yang berharga (EC$process ), kita memerlukan diagram untuk menunjukkan kepada kita informasi lebih rinci tentang rangkaian jalur khusus ini.Namun muncul masalah dari penyajian angka-angka tersebut: terkadang sulit untuk ditafsirkanzscore Informasi disediakan.Bagaimanapun, metode penghitungan ini tidak bersifat universal. Seperti yang ditunjukkan di atas, ini hanyalah jumlah gen yang terregulasi ke atas dikurangi jumlah gen yang terregulasi ke bawah dibagi dengan akar kuadrat dari jumlah gen di setiap jalur, menggunakan metode ini.GOCircleGrafik yang dihasilkan juga menekankan fakta ini.
Lingkaran luar diagram lingkaran menunjukkan nilai logFC gen setiap jalur sebagai titik-titik yang tersebar. Lingkaran merah menunjukkan peningkatan regulasi dan lingkaran biru menunjukkan penurunan regulasi.Parameter dapat digunakanlfc.col Ganti warna. Hal ini juga menjelaskan mengapa dalam beberapa kasus, jalur yang sangat penting memiliki skor z yang mendekati nol. Skor z nol bukan berarti saluran tersebut tidak penting. Hal ini hanya menunjukkan bahwa zscore merupakan ukuran kasar, karena jelas zscore juga tidak memperhitungkan tingkat fungsional dan ketergantungan aktivasi gen individu dalam proses biologis.
GOCircle(circ)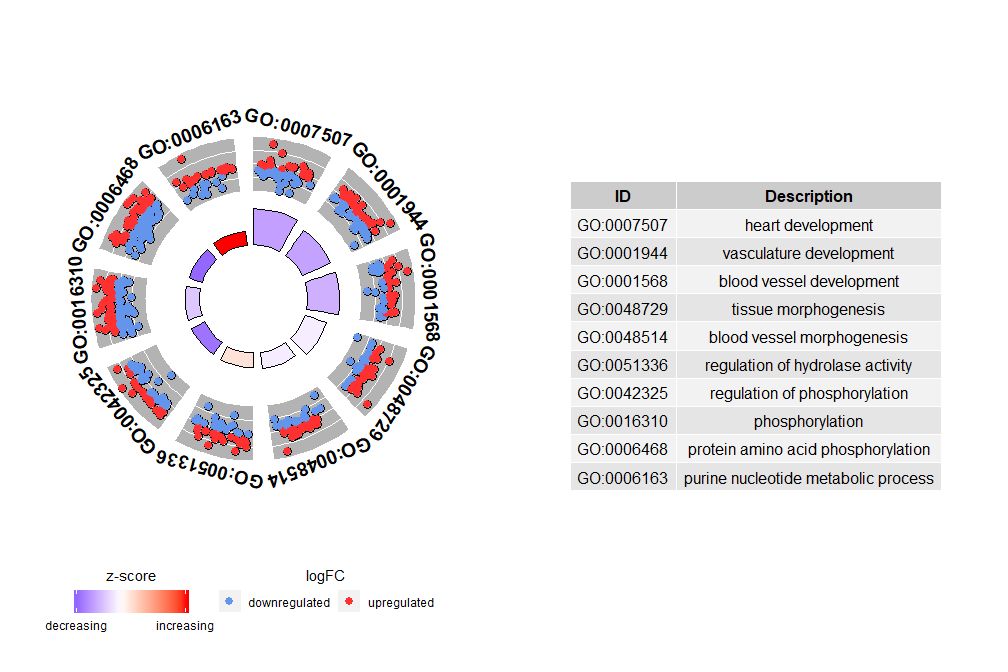
nsub Parameter dapat berupa himpunan angka atau vektor karakter. Jika berupa vektor karakter, maka berisi GO ID atau jalur yang akan ditampilkan;
- # 生成特定通路的圈图
- IDs <- c('GO:0007507', 'GO:0001568', 'GO:0001944', 'GO:0048729', 'GO:0048514', 'GO:0005886', 'GO:0008092', 'GO:0008047')
- GOCircle(circ, nsub = IDs)
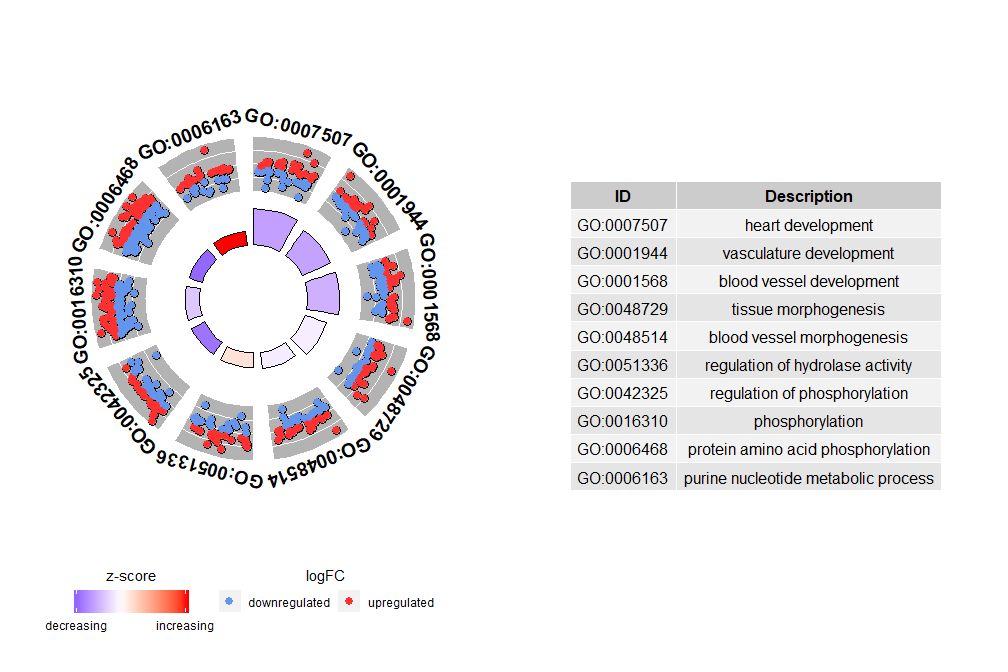
Jika nsub adalah vektor numerik, angka tersebut menentukan angka yang akan ditampilkan. Ini dimulai dari baris pertama bingkai data masukan. Visualisasi ini hanya berfungsi dengan data yang lebih kecil. Jumlah maksimum saluran defaultnya adalah 12. Meskipun jumlah saluran berkurang, jumlah informasi yang ditampilkan bertambah.
- # 圈图展示数据前十个通路
- GOCircle(circ, nsub = 10)
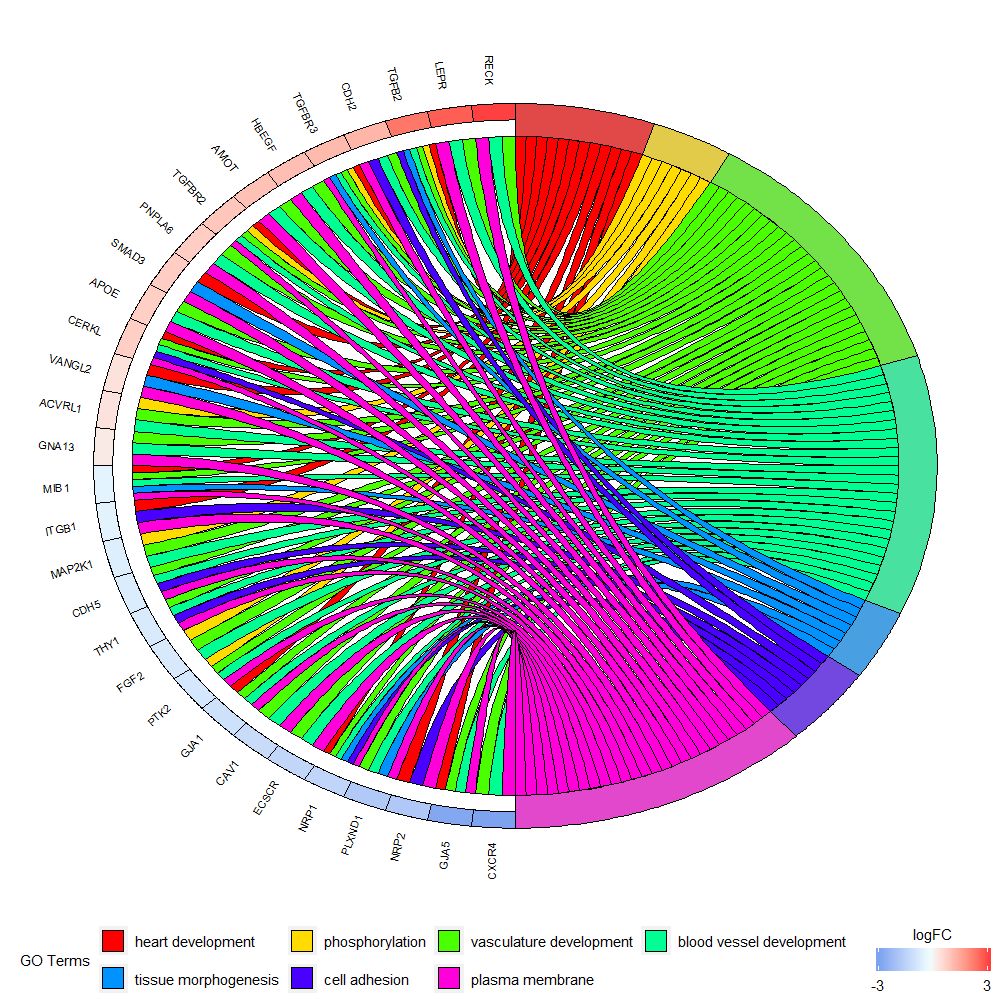
GOCHord dapat menampilkan hubungan antara gen dan jalur yang dipilih serta logFC gen tersebut.Pertama, Anda perlu memasukkan matriks, yang dapat Anda buat sendiri0-1Matriks, Anda juga bisa menggunakan fungsichord_dat Membangun. Fungsi ini memiliki tiga parameter: data, gen, dan proses, dimana dua parameter terakhir harus memiliki setidaknya satu parameter.Lalu fungsinyacircle_datGabungkan data ekspresi dengan hasil analisis fungsional.
Bagan batang dan bagan gelembung dapat memberi Anda kesan pertama terhadap data. Sekarang, Anda dapat memilih beberapa gen dan jalur yang menurut kami berharga. Meskipun GOCircle menambahkan lapisan untuk menampilkan nilai ekspresi gen dalam jalur, GOCircle tidak memiliki informasi individual hubungan antara gen dan berbagai jalur. Tidak mudah untuk mengetahui apakah gen tertentu terkait dengan berbagai proses. GOCHord menutupi kekurangan GOCircle. Baris data yang dihasilkan adalah gen dan kolomnya adalah jalur. "0" berarti gen tersebut tidak ditugaskan ke jalur tersebut, dan "1" adalah kebalikannya.
- # 找到感兴趣的的基因,这里我们以EC$genes为例
- head(EC$genes)
-
- ## ID logFC
- ## 1 PTK2 -0.6527904
- ## 2 GNA13 0.3711599
- ## 3 LEPR 2.6539788
- ## 4 APOE 0.8698346
- ## 5 CXCR4 -2.5647537
- ## 6 RECK 3.6926860
-
- # 获得感兴趣基因的通路
- EC$process
-
- ## [1] "heart development" "phosphorylation"
- ## [3] "vasculature development" "blood vessel development"
- ## [5] "tissue morphogenesis" "cell adhesion"
- ## [7] "plasma membrane"
-
- # 使用chord_dat构建矩阵
- chord <- chord_dat(circ, EC$genes, EC$process)
- head(chord)
-
- ## heart development phosphorylation vasculature development
- ## PTK2 0 1 1
- ## GNA13 0 0 1
- ## LEPR 0 0 1
- ## APOE 0 0 1
- ## CXCR4 0 0 1
- ## RECK 0 0 1
- ## blood vessel development tissue morphogenesis cell adhesion
- ## PTK2 1 0 0
- ## GNA13 1 0 0
- ## LEPR 1 0 0
- ## APOE 1 0 0
- ## CXCR4 1 0 0
- ## RECK 1 0 0
- ## plasma membrane logFC
- ## PTK2 1 -0.6527904
- ## GNA13 1 0.3711599
- ## LEPR 1 2.6539788
- ## APOE 1 0.8698346
- ## CXCR4 1 -2.5647537
- ## RECK 1 3.6926860
Dalam contoh ini, kita melewati dua parameter. Jika hanya parameter gen yang ditentukan, hasilnya adalah daftar gen yang dipilih dan semua konstruksi proses dengan setidaknya satu gen tertentu.0-1matriks; jika hanya ditentukanprocessparameter, hasilnya semua gen menghasilkan0-1 Matriks gen yang ditugaskan pada setidaknya satu proses dalam daftar. Perlu diketahui bahwa hanya menentukan gen dan parameter proses saja dapat menghasilkan matriks 0-1 yang sangat besar, sehingga menghasilkan hasil visualisasi yang membingungkan.
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
-
- # Generate the matrix with a list of selected genes
- chord_genes <- chord_dat(data = circ, genes = EC$genes)
- head(chord_genes)
-
- ## heart development vasculature development blood vessel development
- ## PTK2 0 1 1
- ## GNA13 0 1 1
- ## LEPR 0 1 1
- ## APOE 0 1 1
- ## CXCR4 0 1 1
Bagan ini dimaksudkan untuk menampilkan subkumpulan data berdimensi tinggi yang lebih kecil. Ada dua parameter utama yang dapat disesuaikan:gene.orderDannlfc . Parameter gen dapat ditentukan sebagai 'logFC', 'abjad', 'tidak ada'. Faktanya, kami biasanya menetapkan parameter gen sebagai logFC; parameter nlfc adalah salah satu parameter terpenting dari fungsi ini, karena dapat menangani bagaimana setiap gen memiliki 0 atau lebih nilai logFC yang disajikan dalam matriks. Oleh karena itu kita harus menentukan parameter untuk menghindari kesalahan.
Misalnya, jika Anda memiliki matriks tanpa nilai logFC, Anda harus menyetelnyanlfc=0 ; Atau lakukan analisis ekspresi diferensial pada gen dalam berbagai kondisi atau batch. Dalam hal ini, setiap gen berisi beberapa nilai logFC, dan nomor kolom nlfc=logFC perlu disetel. Nilai defaultnya adalah "1" karena diyakini bahwa seringkali hanya ada satu nilai logFC per gen. Gunakan parameter spasi untuk menentukan jarak antara persegi panjang berwarna yang mewakili logFC. Parameter gene.size menentukan ukuran font nama gen, dan gene.space menentukan ukuran spasi antar nama gen.
- chord <- chord_dat(data = circ, genes = EC$genes, process = EC$process)
- GOChord(chord, space = 0.02, gene.order = 'logFC', gene.space = 0.25, gene.size = 5)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
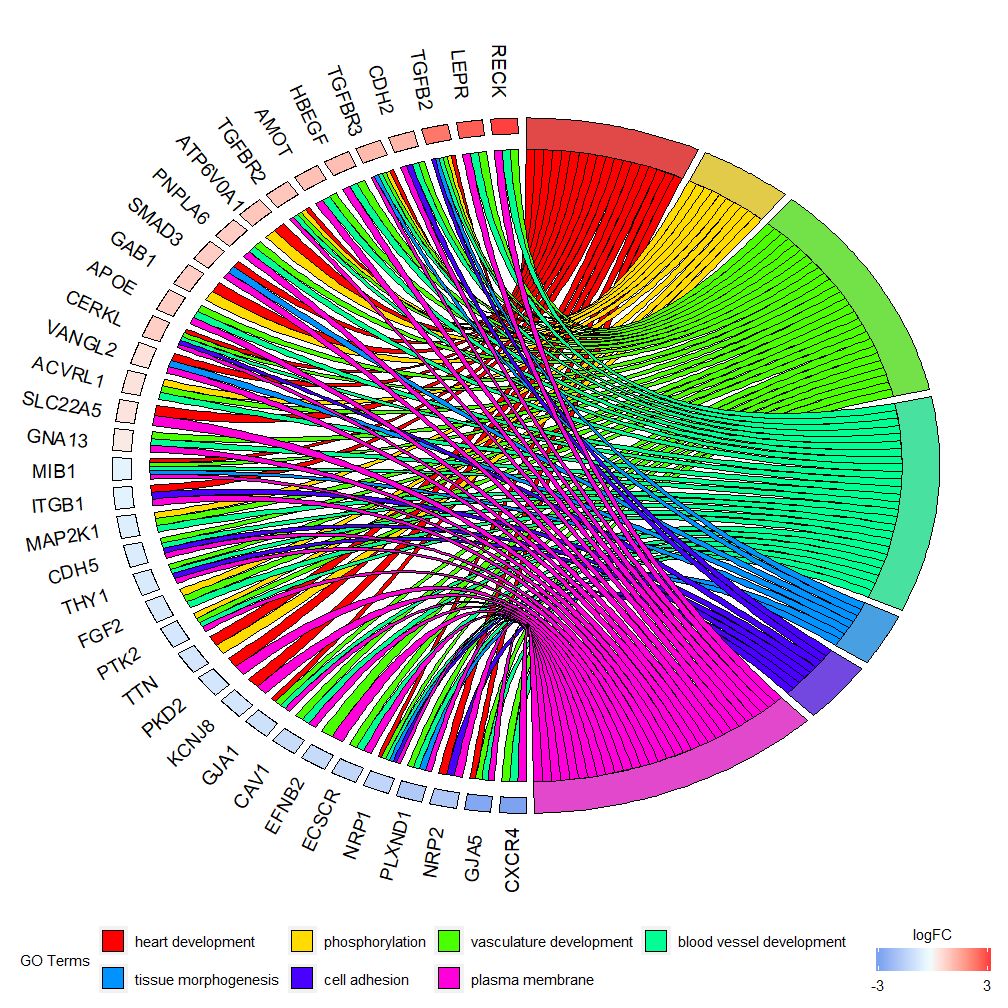
Dapat diatur sesuai dengan nilai logFCgene.order=‘logFC’ , urutkan gen berdasarkan nilai logFC-nya. Terkadang gambar menjadi agak ramai dan hal ini dapat diotomatisasi dengan menggunakan parameter batas untuk mengurangi jumlah gen atau jalur yang ditampilkan. Limit adalah vektor dengan dua nilai cutoff (defaultnya adalah c(0,0)). Nilai pertama menentukan jumlah minimum jalur yang harus ditetapkan gennya. Nilai kedua menentukan jumlah gen yang ditugaskan pada jalur tersebut.
- # 仅显示分配给至少三个通路的基因
- GOChord(chord, limit = c(3, 0), gene.order = 'logFC')
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
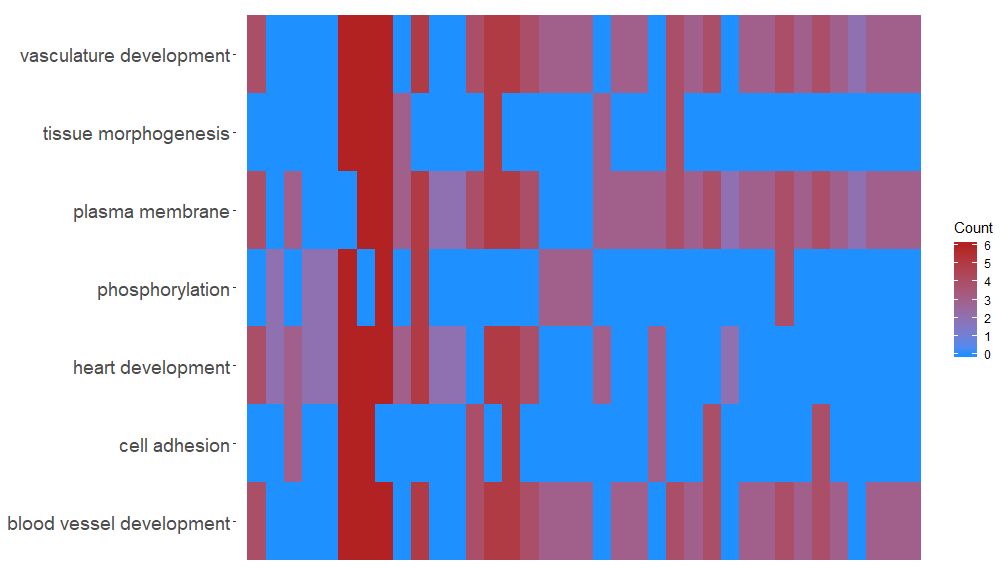
Fungsi GOHeat dapat menampilkan hubungan antara gen dan jalur menggunakan peta panas, mirip dengan GOCHord. Proses biologis ditampilkan secara horizontal dan gen ditampilkan secara vertikal. Setiap kolom dibagi menjadi persegi panjang kecil, dan warnanya umumnya bergantung pada nilai logFC. Selain itu, gen yang diperkaya dalam jalur fungsional serupa juga dikelompokkan. Ada dua mode untuk pemilihan warna peta panas, bergantung pada parameter nlfc. Jika nlfc = 0, warna adalah jumlah jalur yang diperkaya untuk setiap gen. Lihat contoh untuk detailnya:
- # First, we use the chord object without logFC column to create the heatmap
- GOHeat(chord[,-8], nlfc = 0)
GOHeat(chord[,-8])Warna sesuai dengan logFC gen jika nlfc = 1
GOHeat(chord, nlfc = 1, fill.col = c('red', 'yellow', 'green'))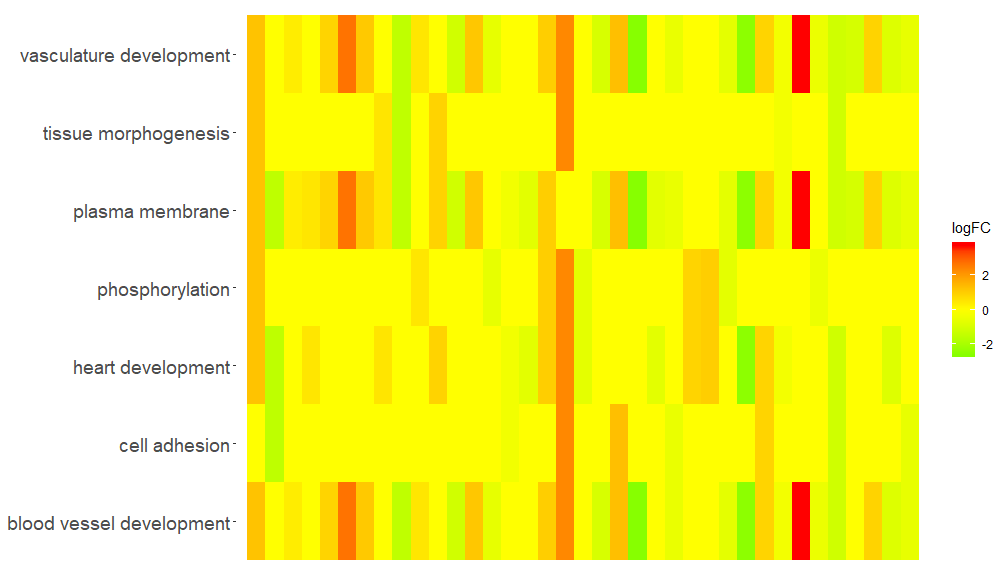
Ide di balik fungsi GOCluster adalah untuk menampilkan informasi sebanyak mungkin. Berikut ini contohnya:
- GOCluster(circ, EC$process, clust.by = 'logFC', term.width = 2)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
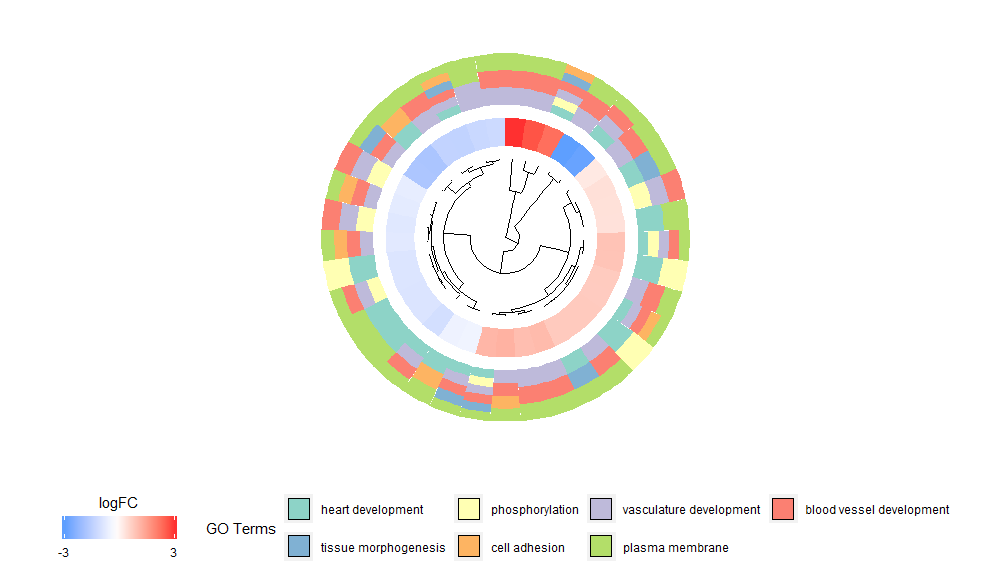
Pengelompokan hierarki adalah metode analisis pengelompokan tanpa pengawasan yang populer untuk ekspresi gen yang memastikan pengelompokan gen yang tidak memihak berdasarkan pola ekspresi, sehingga klaster yang mengelompok bersama mungkin berisi beberapa kelompok gen yang terkoordinasi atau terkait secara fungsional. GOCluster menggunakanhclust Metode melakukan pengelompokan hierarki profil ekspresi gen. Jika ingin mengubah metrik jarak atau algoritma clustering, gunakan parameter metric dan clust masing-masing. Dendrogram yang dihasilkan dapat dikonversi dengan bantuan ggdendro dan divisualisasikan dengan ggplot2. Pilih tata letak melingkar karena tidak hanya efektif tetapi juga menarik secara visual. Lingkaran pertama di sebelah dendrogram mewakili logFC gen, yang sebenarnya adalah daun dari pohon pengelompokan. Jika Anda tertarik dengan beberapa kontras, Anda dapat memodifikasi parameter nlfc, secara default disetel ke "1" sehingga hanya satu cincin yang digambar. Nilai logFC diberi kode warna menggunakan skala warna yang dapat ditentukan pengguna (lfc.col); lingkaran berikutnya mewakili jalur yang ditetapkan ke gen. Agar terlihat bagus, jumlah saluran telah dikurangi, dan warna saluran dapat diubah menggunakan parameter term.col.masih ada?GOCluster untuk melihat cara mengubah parameter. Parameter terpenting dari fungsi ini adalah cluster.by, yang dapat ditentukan ke cluster berdasarkan pola ekspresi gen ('logFC', seperti yang ditunjukkan di atas) atau kategori fungsional ('istilah').
- GOCluster(circ, EC$process, clust.by = 'term', lfc.col = c('darkgoldenrod1', 'black', 'cyan1'))
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
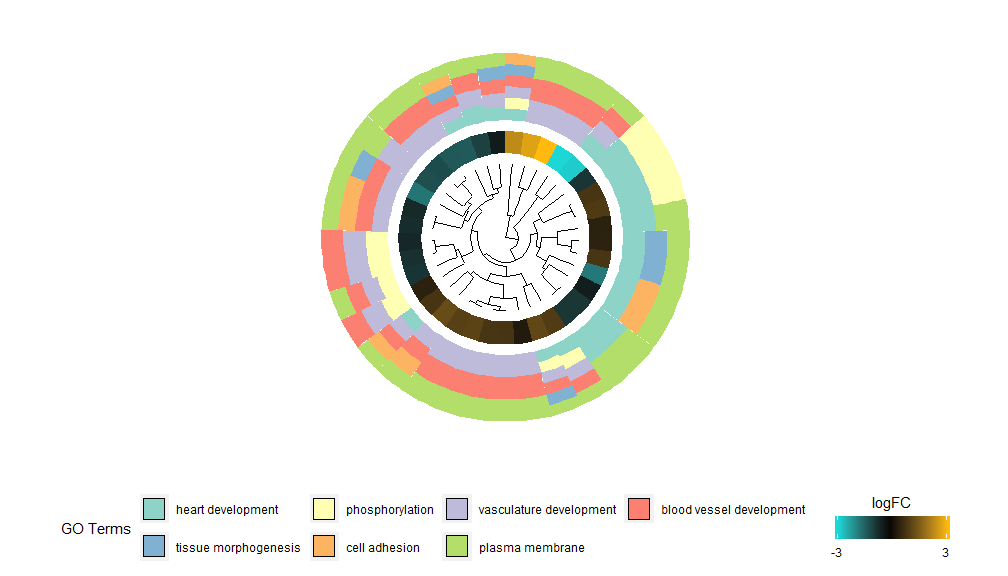
Diagram Venn dapat digunakan untuk mendeteksi hubungan antara berbagai daftar gen yang diekspresikan secara berbeda, atau untuk mengeksplorasi perpotongan beberapa jalur gen dalam analisis fungsional. Diagram Venn tidak hanya menunjukkan jumlah gen yang tumpang tindih, tetapi juga informasi tentang pola ekspresi gen (biasanya up-regulated, sering down-regulated, atau counter-regulated). Saat ini, maksimal tiga kumpulan data digunakan sebagai masukan. Data masukan berisi setidaknya dua kolom: satu untuk nama gen dan satu lagi untuk nilai logFC.
- l1 <- subset(circ, term == 'heart development', c(genes,logFC))
- l2 <- subset(circ, term == 'plasma membrane', c(genes,logFC))
- l3 <- subset(circ, term == 'tissue morphogenesis', c(genes,logFC))
- GOVenn(l1,l2,l3, label = c('heart development', 'plasma membrane', 'tissue morphogenesis'))
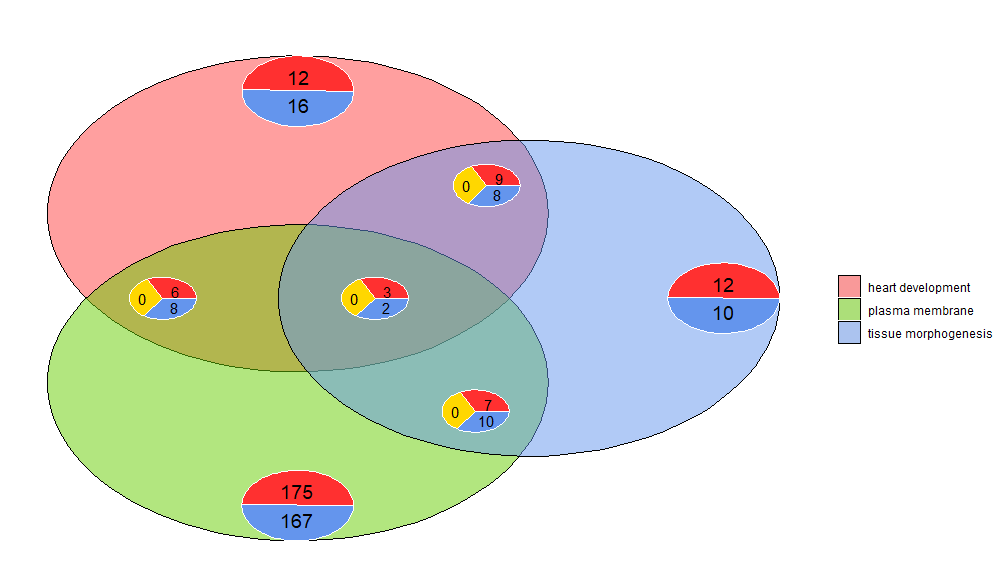
Misalnya, perkembangan jantung dan morfogenesis jaringan memiliki 22 gen, 12 diregulasi ke atas dan 10 diregulasi ke bawah. Hal penting yang perlu diperhatikan adalah diagram lingkaran tidak menampilkan informasi yang berlebihan. Oleh karena itu, jika tiga kumpulan data dibandingkan, gen-gen yang umum pada semua kumpulan data (diagram lingkaran tengah) tidak disertakan dalam diagram lingkaran lainnya. Alat ini tersedia di glossyapp https://wwalter.shinyapps.io/Venn/, alat web lebih interaktif, lingkaran memiliki luas sebanding dengan jumlah gen dalam dataset, dan penggeser dapat digunakan untuk memindahkan diagram lingkaran kecil, dan memiliki Fitur GOVenn semua opsi untuk mengubah tata letak plot dan juga untuk mengunduh gambar dan daftar gen.
Halaman beranda perangkat lunak: https://wencke.github.io/

Ia telah mengabdikan dirinya untuk meneliti teknologi selama lebih dari 30 tahun, dan mahir dalam berbagai bahasa seperti java, linux, javascript, php, css, dll. "Saya telah memberikan banyak kontribusi di bidang dokumentasi pengembang stasiun sumber terbuka untuk berbagi beberapa masalah dalam pengembangan teknologi untuk referensi di masa mendatang. Semua orang memeriksanya

Surat[email protected]