
τα στοιχεία επικοινωνίας μου
Ταχυδρομείο[email protected]
2024-07-12
한어Русский языкEnglishFrançaisIndonesianSanskrit日本語DeutschPortuguêsΕλληνικάespañolItalianoSuomalainenLatina
Το πακέτο GOPlot χρησιμοποιείται για την οπτικοποίηση βιολογικών δεδομένων. Μάλλον, αυτό το πακέτο ενσωματώνει και οπτικοποιεί δεδομένα έκφρασης με τα αποτελέσματα λειτουργικών αναλύσεων.Αλλά πρόσεχεΑυτό το πακέτο δεν μπορεί να χρησιμοποιηθεί για την εκτέλεση αυτών των αναλύσεων, μόνο για την οπτικοποίηση των αποτελεσμάτων. . Σε όλους τους τομείς της επιστήμης, είναι δύσκολο να περιγραφούν ρεαλιστικά τα πράγματα λόγω των περιορισμών χώρου και της απλότητας που απαιτείται για τα αποτελέσματα, επομένως οι πληροφορίες πρέπει να οπτικοποιούνται και οι εικόνες να χρησιμοποιούνται για τη μετάδοση πληροφοριών. Τα καλά σχεδιασμένα γραφικά παρέχουν περισσότερες πληροφορίες σε λιγότερο χώρο. Η ιδέα του πακέτου είναι να επιτρέπει στους χρήστες να εξετάζουν γρήγορα μεγάλες ποσότητες δεδομένων, να αποκαλύπτουν τάσεις στα δεδομένα και να βρίσκουν μοτίβα και συσχετισμούς στα δεδομένα.
Η οπτικοποίηση δεδομένων μπορεί να μας βοηθήσει να βρούμε απαντήσεις σε βιολογικά ερωτήματα, να κρίνουμε μια συγκεκριμένη υπόθεση και ακόμη και να ανακαλύψουμε διαφορετικές γωνίες για τη διερεύνηση διαφορετικών προβλημάτων. Και οι συναρτήσεις σχεδίασης αυτού του πακέτου αναπτύσσονται με βάση την ιεραρχική δομή των δεδομένων, ξεκινώντας από τα συνολικά δεδομένα και τελειώνοντας με ένα υποσύνολο επιλεγμένων γονιδίων και αντίστοιχων μονοπατιών.
Ας το εξηγήσουμε συγκεκριμένα με ένα παράδειγμα.
Ονομάζουμε τα δεδομένα που συνοδεύουν το GOplot, το οποίο προέρχεται από το GEOGSE47067, που περιέχει πληροφορίες μεταγραφής ενδοθηλιακών κυττάρων από δύο ιστούς (εγκέφαλο και καρδιά για περισσότερες πληροφορίες, δείτε την εργασία των Nolan et al.Τα δεδομένα κανονικοποιούνται και εντοπίζονται γονίδια που εκφράζονται με διαφορετικό τρόπο.και, στη συνέχεια, χρησιμοποιήστε το εργαλείο σχολιασμού συνάρτησης DAVID (τα δεδομένα σχολιασμού DAVID ενημερώνονται αργά και δεν συνιστάται τώρα. Συνιστάται η χρήσηGo East, το καλύτερο διαδικτυακό εργαλείο ανάλυσης εμπλουτισμού GOκαιΑυτός ο ιστότοπος, ο οποίος μπορεί να πραγματοποιήσει ανάλυση εμπλουτισμού σε ένα μόνο βήμα, έχει αναφερθεί περισσότερες από 350 φορές από το CNS και άλλους πριν από τη δημοσίευσή του.Εκτελέστε ανάλυση εμπλουτισμού,Master GSEA σε ένα άρθρο, εξαιρετικά λεπτομερές σεμινάριο) Γονιδιακός σχολιασμός γονιδίων διαφορικής έκφρασης (adjusted p-value < 0.05 ) και ανάλυση λειτουργικού εμπλουτισμού. Αυτό το σύνολο δεδομένων περιέχει τις ακόλουθες πέντε κατηγορίες δεδομένων:
| όνομα | περιγράφω | Μέγεθος συνόλου δεδομένων |
|---|---|---|
| EC$set | Ομαλοποιημένη γονιδιακή έκφραση σε ενδοθηλιακά κύτταρα εγκεφάλου και καρδιάς (3 επαναλήψεις) | 20644 x 7 |
| EC$genelist | Γονίδια με διαφορική έκφραση (προσαρμοσμένη τιμή p < 0,05) | 2039 x 7 |
| EC$david | Αποτελέσματα ανάλυσης λειτουργικού εμπλουτισμού διαφορικών γονιδίων με χρήση DAVID | 174 x 5 |
| EC$γονίδιο | Γονίδια και logFC | 37 x 2 |
| διαδικασία EC$ | Επιλεγμένοι φορείς χαρακτηριστικών για εμπλουτισμένες βιολογικές διεργασίες | 7 |
Θέλουμε να δούμε τις εμπλουτισμένες με GO μονοπάτια των διαφορικά εκφραζόμενων γονιδίων, αλλά πριν αρχίσουμε να σχεδιάζουμε πρέπει να παρέχουμε δεδομένα που να πληρούν τις απαιτήσεις μορφής.Σε γενικές γραμμές, τα δεδομένα που απαιτούνται για τη σχεδίαση του γραφήματος παρέχονται από εσάς, αλλάΥπάρχει μια λειτουργία σε αυτό το πακέτοcircle_datΜπορεί να μας βοηθήσει να αντιμετωπίσουμε τη μορφή δεδομένων。circle_datΜπορεί να συνδυάσει τα αποτελέσματα ανάλυσης λειτουργικού εμπλουτισμού επιλεγμένων γονιδίων και τις τιμές logFC τους, κυρίως για διαφορικά εκφραζόμενα γονίδια.circle_dat Η χρήση είναι πολύ απλή, απλά διαβάστε σε δύο δεδομένα. Τα πρώτα δεδομένα περιέχουν τα αποτελέσματα της ανάλυσης λειτουργικού εμπλουτισμού, με τουλάχιστον τέσσερις στήλες (κατηγορία ανάλυσης λειτουργικού εμπλουτισμού, διαδρομή, γονίδιο, προσαρμοσμένη τιμή p).Τα δεύτερα δεδομένα είναι του επιλεγμένου γονιδίου και του logFC του, αυτά τα δεδομένα μπορεί να είναι η πηγήlimmaΤα αποτελέσματα της στατιστικής ανάλυσης (Σημείωση από Βιογραφίες: Φροντίστε να δώσετε προσοχή σε δύο αρχείαΠώς ονομάζονται τα γονίδιαΝα είστε συνεπείς, όπως όλοιGene symbol ). Ας χρησιμοποιήσουμε παραδείγματα για να δούμε τις μορφές δεδομένων που αναφέρονται παραπάνω.
- #安装已发布的稳定版本
- #install.packages('GOplot')
- #安装github上的开发版本
- #install_github('wencke/wencke.github.io')
- #载入包
- library(GOplot)
- #读入包内自带的数据
- data(EC)
- #查看功能富集分析结果的数据格式
- head(EC$david)
-
- ## Category ID Term
- ## 1 BP GO:0007507 heart development
- ## 2 BP GO:0001944 vasculature development
- ## 3 BP GO:0001568 blood vessel development
- ## 4 BP GO:0048729 tissue morphogenesis
- ## 5 BP GO:0048514 blood vessel morphogenesis
- ## 6 BP GO:0051336 regulation of hydrolase activity
- ## Genes
- ## 1 DLC1, NRP2, NRP1, EDN1, PDLIM3, GJA1, TTN, GJA5, ZIC3, TGFB2, CERKL, GATA6, COL4A3BP, GAB1, SEMA3C, MKL2, SLC22A5, MB, PTPRJ, RXRA, VANGL2, MYH6, TNNT2, HHEX, MURC, MIB1, FOXC2, FOXC1, ADAM19, MYL2, TCAP, EGLN1, SOX9, ITGB1, CHD7, HEXIM1, PKD2, NFATC4, PCSK5, ACTC1, TGFBR2, NF1, HSPG2, SMAD3, TBX1, TNNI3, CSRP3, FOXP1, KCNJ8, PLN, TSC2, ATP6V0A1, TGFBR3, HDAC9
- ## 2 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, EFNB2, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 3 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 4 DLC1, ENAH, NRP1, PGF, ZIC2, TGFB2, CD44, ILK, SEMA3C, RET, AR, RXRA, VANGL2, LEF1, TNNT2, HHEX, MIB1, NCOA3, FOXC2, FOXC1, TGFB1I1, WNT5A, COBL, BBS4, FGFR3, TNC, BMPR2, CTNND1, EGLN1, NR3C1, SOX9, TCF7L1, IGF1R, FOXQ1, MACF1, HOXA5, BCL2, PLXND1, CAR2, ACTC1, TBX4, SMAD3, FZD3, SHANK3, FZD6, HOXB4, FREM2, TSC2, ZIC5, TGFBR3, APAF1
- ## 5 GNA13, CAV1, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, CDH2, MEIS1, WT1, TGFB2, WARS, PTK2, CERKL, APOE, CXCR4, ANG, SEMA3C, PLCD1, NOS2, MKL2, PLXND1, FIGF, FGF2, PTPRJ, TGFBR2, TBX4, NF1, TBX1, TNNI3, PNPLA6, VASH1, THY1, NUS1, MEOX2, ECSCR, AMOT, HBEGF, FOXC2, FOXC1, HDAC7
- ## 6 CAV1, XIAP, AGFG1, ADORA2A, TNNC1, TBC1D9, LEPR, ABHD5, EDN1, ASAP2, ASAP3, SMAP1, TBC1D12, ANG, TBC1D14, MTCH1, TBC1D13, TBC1D4, TBC1D30, DHCR24, HIP1, VAV3, NOS1, NF1, MYH6, RICTOR, TBC1D22A, THY1, PLCE1, RNF7, NDEL1, CHML, IFT57, ACAP2, TSC2, ERN1, APAF1, ARAP3, ARAP2, ARAP1, HTR2A, F2R
- ## adj_pval
- ## 1 0.000002170
- ## 2 0.000010400
- ## 3 0.000007620
- ## 4 0.000119000
- ## 5 0.000720000
- ## 6 0.001171166
-
- #查看基因的数据格式
- head(EC$genelist)
-
- ## ID logFC AveExpr t P.Value adj.P.Val B
- ## 1 Slco1a4 6.645388 1.2168670 88.65515 1.32e-18 2.73e-14 29.02715
- ## 2 Slc19a3 6.281525 1.1600468 69.95094 2.41e-17 2.49e-13 27.62917
- ## 3 Ddc 4.483338 0.8365231 65.57836 5.31e-17 3.65e-13 27.18476
- ## 4 Slco1c1 6.469384 1.3558865 59.87613 1.62e-16 8.34e-13 26.51242
- ## 5 Sema3c 5.515630 2.3252117 58.53141 2.14e-16 8.81e-13 26.33626
- ## 6 Slc38a3 4.761755 0.9218670 54.11559 5.58e-16 1.76e-12 25.70308
Αφού κατανοήσετε τις δύο μορφές δεδομένων εισόδου, μπορείτε να χρησιμοποιήσετεcirlce_datλειτουργία για τη δημιουργία δεδομένων σχεδίασης.
- # 生成画图所需的数据格式
- circ <- circle_dat(EC$david, EC$genelist)
-
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
circΤο αντικείμενο έχει οκτώ στήλες δεδομένων, δηλαδή
Κατηγορία: BP (Βιολογική διεργασία), CC (Κυτταρικό Συστατικό) ή MF (Μοριακή λειτουργία)
Αναγνωριστικό: GO id (προαιρετική στήλη, εάν θέλετε να χρησιμοποιήσετε ένα εργαλείο λειτουργικής ανάλυσης που δεν βασίζεται στο αναγνωριστικό GO, δεν μπορείτε να επιλέξετε τη στήλη ID. Το αναγνωριστικό εδώ μπορεί επίσης να είναι αναγνωριστικό KEGG)
όρος: GO μονοπάτι
καταμέτρηση: αριθμός γονιδίων σε κάθε μονοπάτι
γονίδιο: όνομα γονιδίου - logFC: τιμή logFC κάθε γονιδίου
adj_pval: προσαρμοσμένη τιμή p, μονοπάτια με adj_pval<0,05 θεωρούνται ότι είναι σημαντικά εμπλουτισμένα
zscore: Το zscore δεν αναφέρεται σε μια μέθοδο στατιστικής κανονικοποίησης, αλλά είναι μια εύκολα υπολογισμένη τιμή για να εκτιμηθεί εάν μια βιολογική διεργασία (/μοριακή λειτουργία/κυτταρικό συστατικό) είναι πιο πιθανό να μειωθεί (αρνητική τιμή) ή να αυξηθεί (θετική τιμή).Η μέθοδος υπολογισμού είναι ο αριθμός των γονιδίων που ρυθμίζονται προς τα πάνω μείον τον αριθμό των γονιδίων που ρυθμίζονται προς τα κάτω με την τετραγωνική ρίζα του αριθμού των γονιδίων σε κάθε μονοπάτι.
Όταν κοιτάμε για πρώτη φορά τα δεδομένα, θέλουμε να δείξουμε όσο το δυνατόν περισσότερα μονοπάτια από το γράφημα και θέλουμε επίσης να βρούμε πολύτιμα μονοπάτια, επομένως χρειαζόμαστε ορισμένες παραμέτρους για να αξιολογήσουμε τη σημασία. Τα γραφήματα ράβδων χρησιμοποιούνται συχνά για να περιγράψουν δείγματα δεδομένων, επομένως μπορούμε να χρησιμοποιήσουμε τη συνάρτηση GObar για να δημιουργήσουμε γρήγορα ένα όμορφο ραβδωτό γράφημα.
Πρώτον, δημιουργείται ένα απλό γράφημα ράβδων Ο οριζόντιος άξονας είναιGO Terms, σύμφωνα με τουςzscoreΤαξινόμηση των ράβδων είναι ο κατακόρυφος άξονας-log(adj p-value);Το χρώμα αντιπροσωπεύειzscore, το μπλε δείχνειz-scoreείναι αρνητική τιμή, η γονιδιακή έκφραση στην αντίστοιχη οδό είναι πιο πιθανό να μειωθεί, που υποδεικνύεται με κόκκινοz-score είναι θετική τιμή, η γονιδιακή έκφραση στην αντίστοιχη οδό είναι πιο πιθανό να αυξηθεί. Εάν θέλετε, η σειρά μπορεί να αλλάξει ορίζοντας την παράμετρο order.by.zscore σε FALSE, οπότε οι ράβδοι ταξινομούνται με βάση τη σημασία τους.
- # 生成简单的条形图
- GOBar(subset(circ, category == 'BP'))
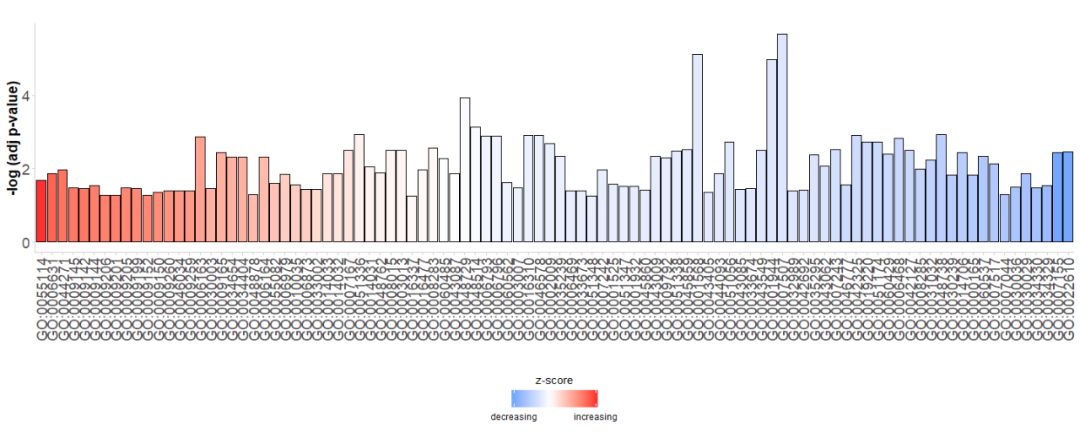
#GOBar(subset(circ, category == 'BP',order.by.zscore=FALSE))Επιπλέον, αλλάξτε την παράμετρο εμφάνισης για να σχεδιάσετε ένα γράφημα ράβδων σύμφωνα με την κατηγορία του καναλιού.
- #根据通路的类别来绘制条形图
- GOBar(circ, display = 'multiple')
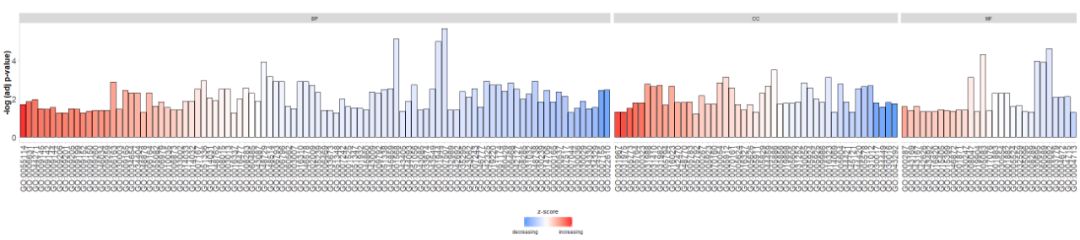
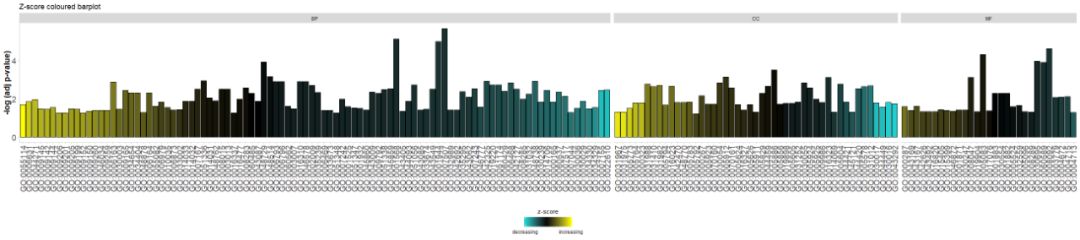
Προσθέστε έναν τίτλο και χρησιμοποιήστε παραμέτρουςzsc.colΑλλαγήzscores χρώμα.
- # Facet the barplot, add a title and change the colour scale for the z-score
- GOBar(circ, display = 'multiple', title = 'Z-score coloured barplot', zsc.col = c('yellow', 'black', 'cyan'))
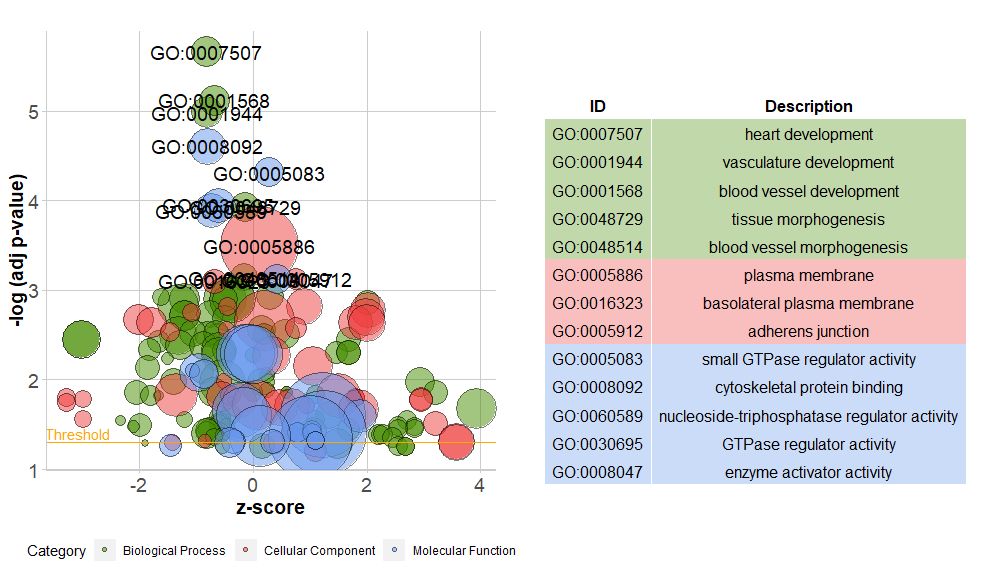
Τα γραφήματα ράβδων είναι πολύ κοινά και εύκολα κατανοητά, αλλά μπορούμε να χρησιμοποιήσουμε γραφήματα με φυσαλίδες για να εμφανίσουμε περισσότερες πληροφορίες σχετικά με τα δεδομένα.
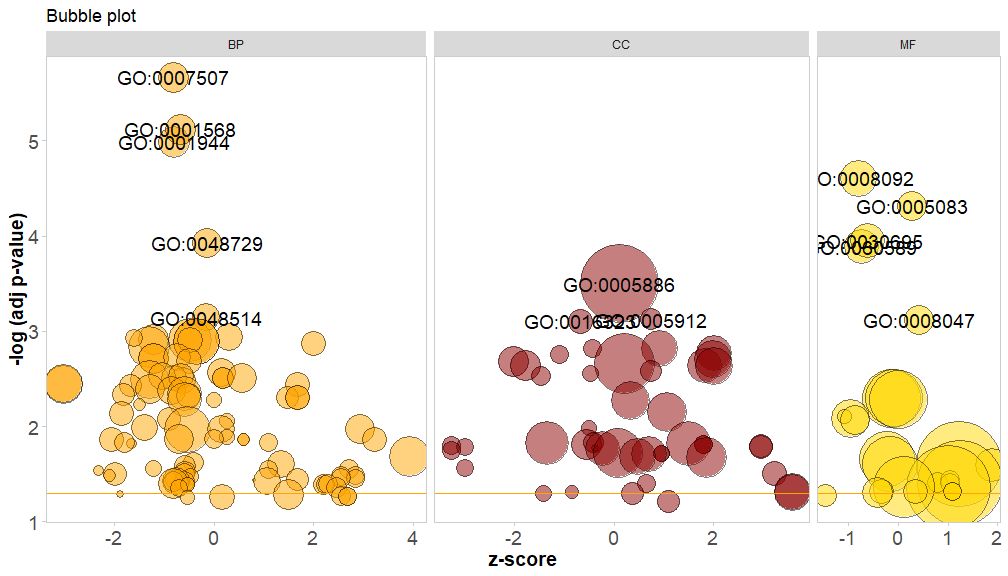
Ο οριζόντιος άξονας είναιzscore;Ο κατακόρυφος άξονας είναι-log(adj p-value), παρόμοιο με ένα διάγραμμα ράβδων, όσο υψηλότερο είναι, τόσο πιο σημαντικός είναι ο εμπλουτισμός του εμβαδού του κύκλου με τον αριθμό των γονιδίων στην αντίστοιχη οδό.circ$count το χρώμα αντιστοιχεί στην κατηγορία που αντιστοιχεί στην οδό, το πράσινο είναι η βιολογική διαδικασία, το κόκκινο είναι το κυτταρικό συστατικό και το μπλε είναι η μοριακή λειτουργία.Μπορεί να εισαχθεί από?GOBubble Δείτε τη σελίδα βοήθειας της συνάρτησης GOBubble για να αλλάξετε όλες τις παραμέτρους της εικόνας. Από προεπιλογή, κάθε κύκλος επισημαίνεται με ένα αντίστοιχο αναγνωριστικό GO και ένας πίνακας που δείχνει την αντίστοιχη σχέση μεταξύ του GO ID και του όρου GO εμφανίζεται επίσης στα δεξιά.Οι παράμετροι μπορούν να ρυθμιστούν απόtable.legendΓιαFALSE να το κρύψει. Εάν θέλετε να εμφανιστεί η περιγραφή της διαδρομής, ορίστε την παράμετρο ID σε FALSE.Ωστόσο, λόγω του περιορισμένου χώρου και των επικαλυπτόμενων κύκλων, δεν επισημαίνονται όλοι οι κύκλοι, μόνο το-log(adj p-value) > 3(η προεπιλογή είναι 5).
- # 生成泡泡图,并展示-log(adj p-value) > 3 的通路的GO ID
- GOBubble(circ, labels = 3)
Εάν θέλετε να προσθέσετε έναν τίτλο στο γράφημα με συννεφάκια ή να καθορίσετε το χρώμα του κύκλου και να εμφανίσετε τις διαδρομές κάθε κατηγορίας ξεχωριστά και να αλλάξετε το εμφανιζόμενο όριο GO ID, μπορείτε να προσθέσετε τις ακόλουθες παραμέτρους:
GOBubble(circ, title = 'Bubble plot', colour = c('orange', 'darkred', 'gold'), display = 'multiple', labels = 3)
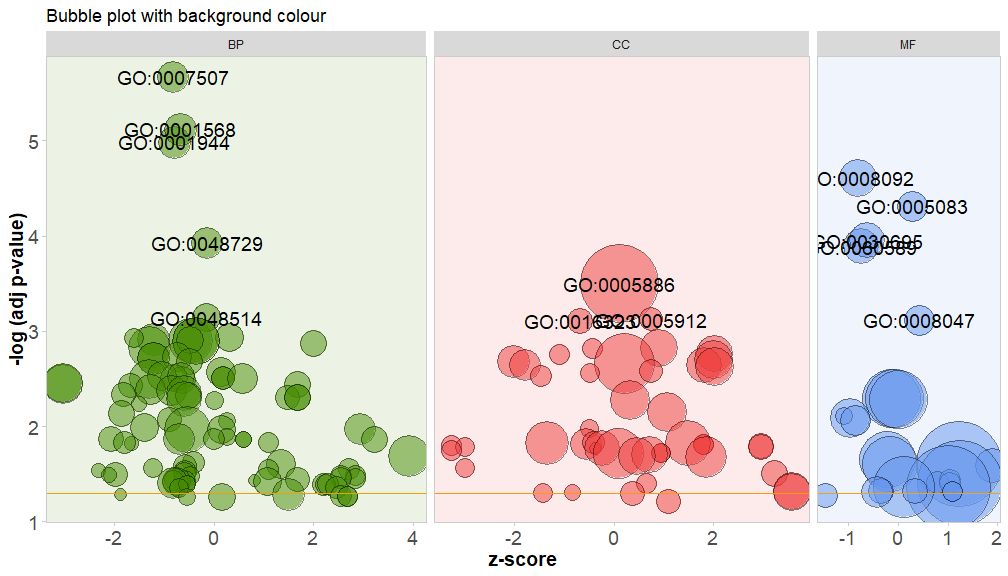
Χρωματίστε το φόντο της κλάσης του καναλιού ορίζοντας την παράμετρο bg.col σε TRUE.
GOBubble(circ, title = 'Bubble plot with background colour', display = 'multiple', bg.col = T, labels = 3)
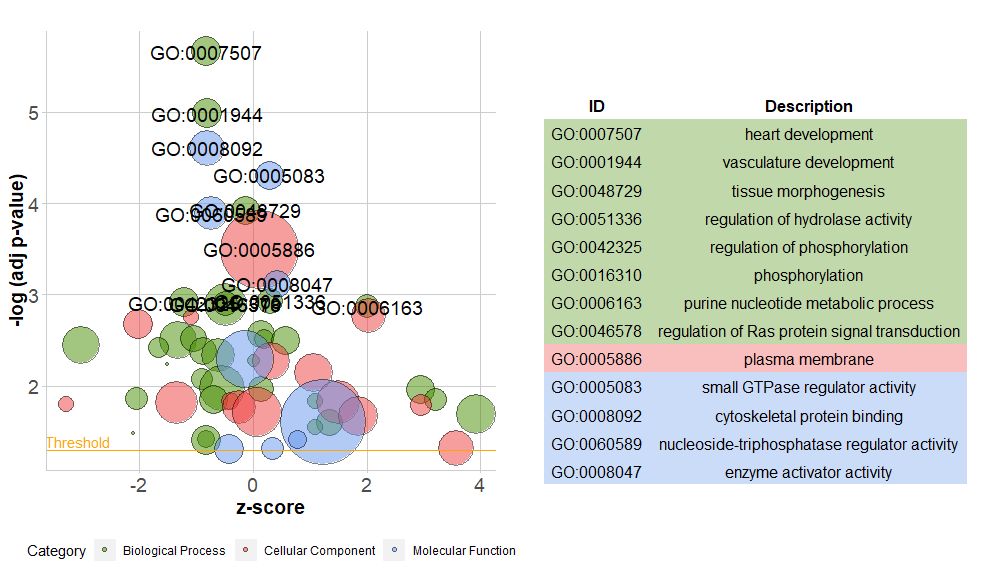
Η νέα έκδοση του πακέτου περιέχει μια νέα λειτουργίαreduce_overlap , αυτή η συνάρτηση μπορεί να μειώσει τον αριθμό των περιττών στοιχείων, δηλαδή, μπορεί να διαγράψει όλες τις οδούς των οποίων η γονιδιακή επικάλυψη είναι μεγαλύτερη ή ίση με το καθορισμένο όριο και να διατηρήσει μόνο ένα μονοπάτι από κάθε ομάδα ως αντιπροσωπευτικό, ανεξάρτητα από την εμφάνιση όλων μονοπάτια στο GO. Με τη μείωση του αριθμού των περιττών όρων, βελτιώνεται σημαντικά η αναγνωσιμότητα των διαγραμμάτων (όπως οι γραφικές παραστάσεις με φυσαλίδες).
- # reduce_overlap,参数设置为0.75
- reduced_circ <- reduce_overlap(circ, overlap = 0.75)
-
- GOBubble(reduced_circ, labels = 2.8)
Αν και ένα γράφημα που δείχνει όλες τις πληροφορίες μπορεί να μας βοηθήσει να ανακαλύψουμε ποια μονοπάτια είναι πιο σημαντικά, η πραγματικότητα εξακολουθεί να εξαρτάται από τις υποθέσεις και τις ιδέες που θέλετε να επιβεβαιώσετε με τα δεδομένα και οι πιο σημαντικές διαδρομές μπορεί να μην είναι απαραίτητα αυτές που σας ενδιαφέρουν. Επομένως, επιλέγοντας με το χέρι ένα πολύτιμο σύνολο μονοπατιών (EC$process ), χρειαζόμαστε ένα διάγραμμα για να μας δείξει πιο λεπτομερείς πληροφορίες σχετικά με αυτό το συγκεκριμένο σύνολο μονοπατιών.Αλλά προκύπτει ένα πρόβλημα από την παρουσίαση αυτών των αριθμών: μερικές φορές είναι δύσκολο να ερμηνευθείzscore Πληροφορίες που παρέχονται.Εξάλλου, αυτή η μέθοδος υπολογισμού δεν είναι καθολική, όπως φαίνεται παραπάνω, είναι απλώς ο αριθμός των γονιδίων που ρυθμίζονται προς τα πάνω μείον τον αριθμό των γονιδίων που ρυθμίζονται προς τα κάτω με την τετραγωνική ρίζα του αριθμού των γονιδίων σε κάθε μονοπάτι.GOCircleΤο γράφημα που προκύπτει τονίζει επίσης αυτό το γεγονός.
Ο εξωτερικός κύκλος του κυκλικού διαγράμματος δείχνει την τιμή logFC των γονιδίων κάθε μονοπατιού ως διάσπαρτα σημεία. Οι κόκκινοι κύκλοι υποδεικνύουν ανοδική ρύθμιση και μπλε υποδηλώνουν μείωση.Μπορούν να χρησιμοποιηθούν παράμετροιlfc.col Αλλαξε χρώμα. Αυτό εξηγεί επίσης γιατί σε ορισμένες περιπτώσεις πολύ σημαντικά μονοπάτια έχουν zscore κοντά στο μηδέν. Ένα zscore μηδέν δεν σημαίνει ότι το κανάλι δεν είναι σημαντικό. Απλώς δείχνει ότι το zscore είναι ένα πρόχειρο μέτρο, γιατί προφανώς το zscore επίσης δεν λαμβάνει υπόψη το λειτουργικό επίπεδο και την εξάρτηση ενεργοποίησης μεμονωμένων γονιδίων σε βιολογικές διεργασίες.
GOCircle(circ)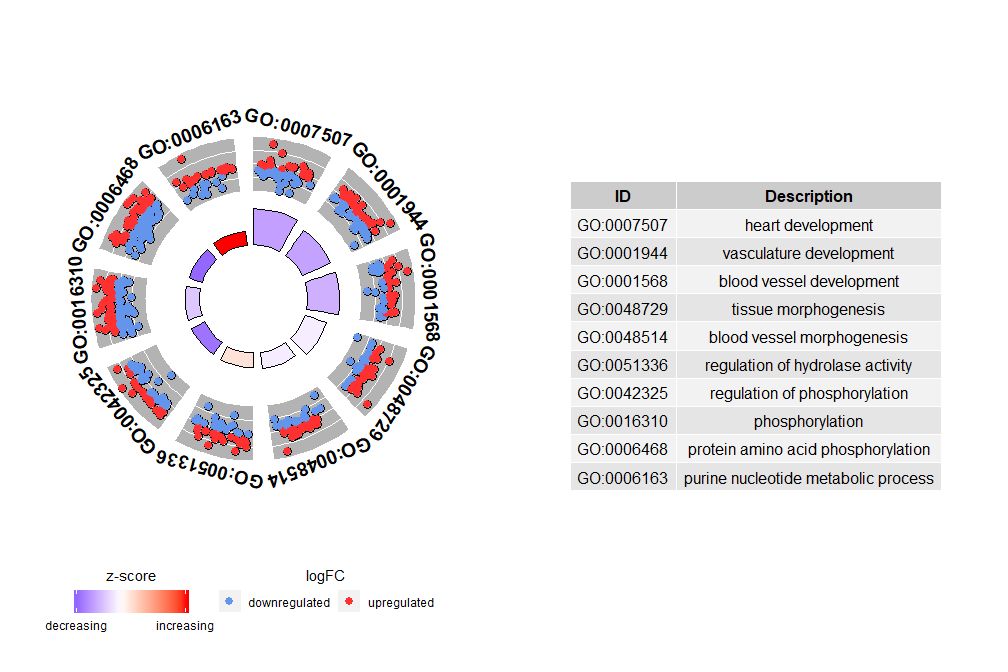
nsub Οι παράμετροι μπορούν να οριστούν αριθμοί ή διανύσματα χαρακτήρων. Εάν είναι διάνυσμα χαρακτήρων, περιέχει το αναγνωριστικό GO ή τη διαδρομή προς εμφάνιση.
- # 生成特定通路的圈图
- IDs <- c('GO:0007507', 'GO:0001568', 'GO:0001944', 'GO:0048729', 'GO:0048514', 'GO:0005886', 'GO:0008092', 'GO:0008047')
- GOCircle(circ, nsub = IDs)
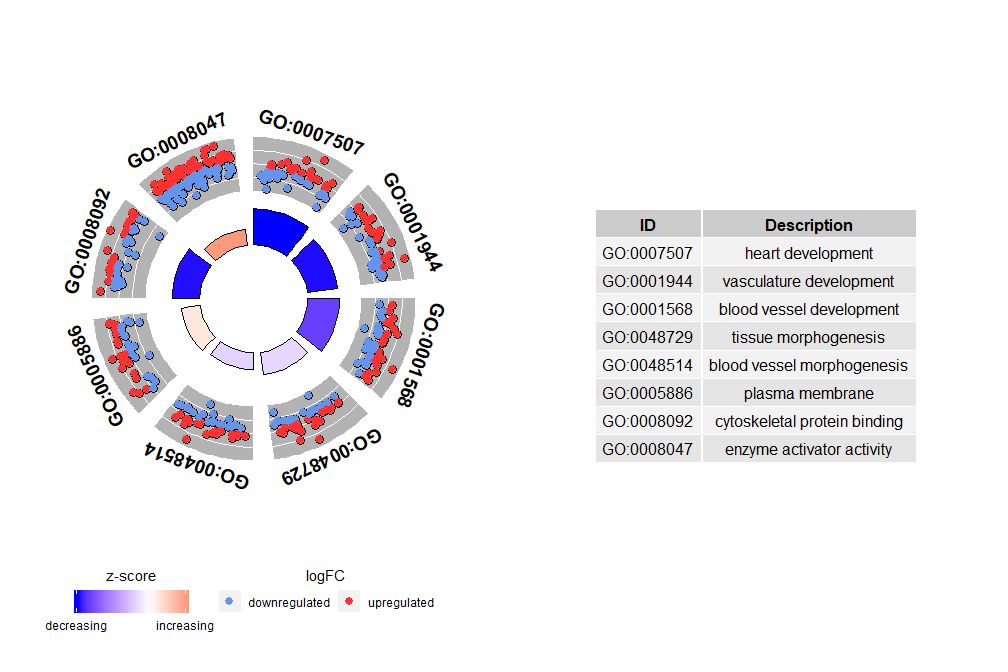
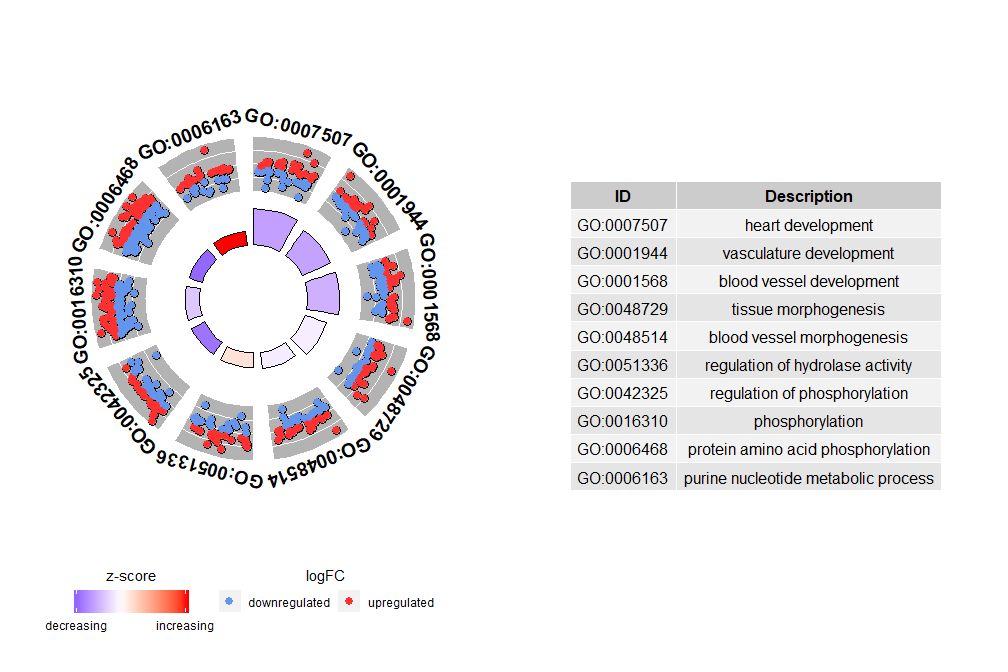
Εάν το nsub είναι αριθμητικό διάνυσμα, ο αριθμός καθορίζει τον αριθμό που θα εμφανιστεί. Ξεκινά από την πρώτη σειρά του πλαισίου δεδομένων εισόδου. Αυτή η οπτικοποίηση λειτουργεί μόνο με μικρότερα δεδομένα. Ο μέγιστος αριθμός καναλιών είναι από προεπιλογή 12. Αν και ο αριθμός των καναλιών μειώνεται, ο όγκος των πληροφοριών που εμφανίζονται αυξάνεται.
- # 圈图展示数据前十个通路
- GOCircle(circ, nsub = 10)
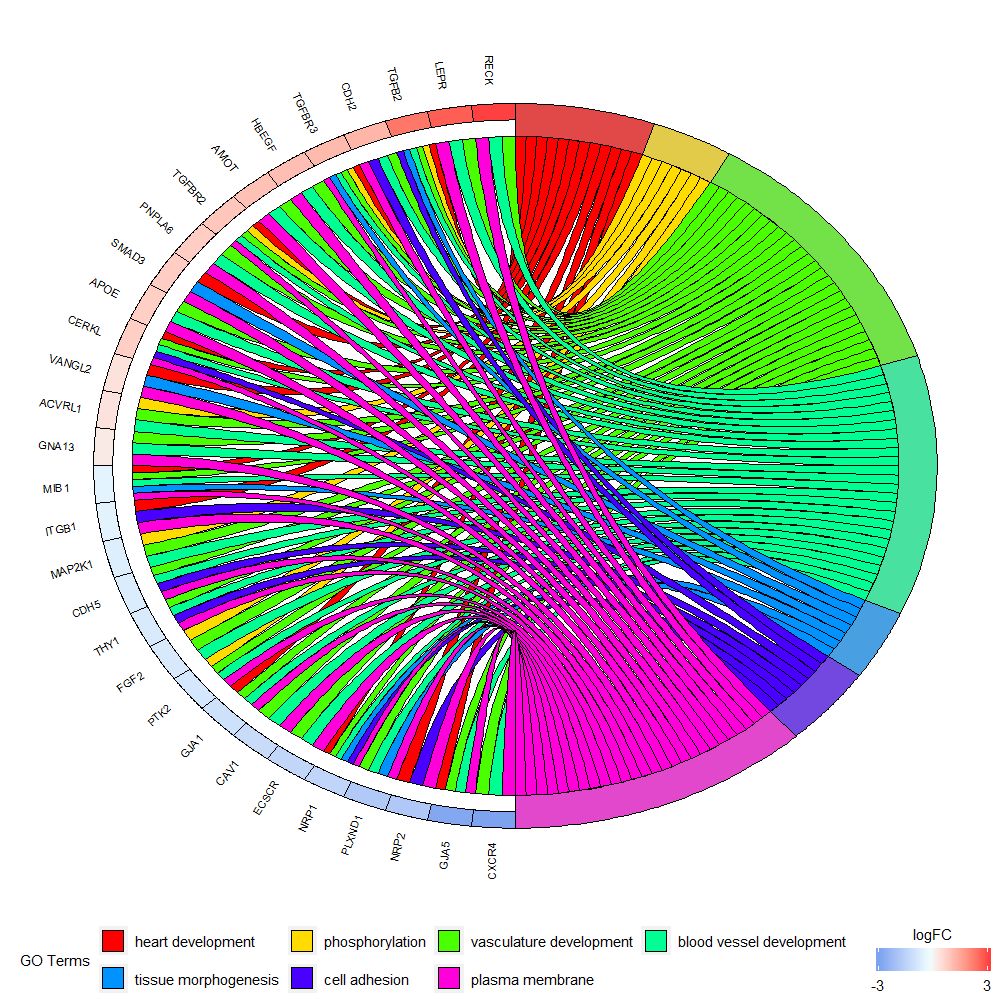
Το GOChord μπορεί να εμφανίσει τη σχέση μεταξύ επιλεγμένων γονιδίων και μονοπατιών και το logFC των γονιδίων.Πρώτα πρέπει να εισάγετε έναν πίνακα, τον οποίο μπορείτε να φτιάξετε μόνοι σας0-1Matrix, μπορείτε επίσης να χρησιμοποιήσετε συναρτήσειςchord_dat Κατασκευάσει. Αυτή η συνάρτηση έχει τρεις παραμέτρους: δεδομένα, γονίδια και διεργασία, εκ των οποίων οι δύο τελευταίες παράμετροι πρέπει να έχουν τουλάχιστον μία παράμετρο.Στη συνέχεια η συνάρτησηcircle_datΣυνδυάστε δεδομένα έκφρασης με αποτελέσματα από λειτουργικές αναλύσεις.
Τα γραφήματα ράβδων και τα γραφήματα με φυσαλίδες μπορούν να σας δώσουν μια πρώτη εντύπωση των δεδομένων Τώρα, μπορείτε να επιλέξετε ορισμένα γονίδια και μονοπάτια που πιστεύουμε ότι είναι πολύτιμα σχετικά με τις σχέσεις μεταξύ γονιδίων και πολλαπλών οδών. Δεν είναι εύκολο να καταλάβουμε εάν ορισμένα γονίδια συνδέονται με πολλαπλές διαδικασίες. Το GOChord αναπληρώνει τις ελλείψεις του GOCircle. Οι σειρές των δεδομένων που δημιουργούνται είναι γονίδια και οι στήλες είναι μονοπάτια "0" σημαίνει ότι το γονίδιο δεν έχει εκχωρηθεί στο μονοπάτι και το "1" είναι το αντίθετο.
- # 找到感兴趣的的基因,这里我们以EC$genes为例
- head(EC$genes)
-
- ## ID logFC
- ## 1 PTK2 -0.6527904
- ## 2 GNA13 0.3711599
- ## 3 LEPR 2.6539788
- ## 4 APOE 0.8698346
- ## 5 CXCR4 -2.5647537
- ## 6 RECK 3.6926860
-
- # 获得感兴趣基因的通路
- EC$process
-
- ## [1] "heart development" "phosphorylation"
- ## [3] "vasculature development" "blood vessel development"
- ## [5] "tissue morphogenesis" "cell adhesion"
- ## [7] "plasma membrane"
-
- # 使用chord_dat构建矩阵
- chord <- chord_dat(circ, EC$genes, EC$process)
- head(chord)
-
- ## heart development phosphorylation vasculature development
- ## PTK2 0 1 1
- ## GNA13 0 0 1
- ## LEPR 0 0 1
- ## APOE 0 0 1
- ## CXCR4 0 0 1
- ## RECK 0 0 1
- ## blood vessel development tissue morphogenesis cell adhesion
- ## PTK2 1 0 0
- ## GNA13 1 0 0
- ## LEPR 1 0 0
- ## APOE 1 0 0
- ## CXCR4 1 0 0
- ## RECK 1 0 0
- ## plasma membrane logFC
- ## PTK2 1 -0.6527904
- ## GNA13 1 0.3711599
- ## LEPR 1 2.6539788
- ## APOE 1 0.8698346
- ## CXCR4 1 -2.5647537
- ## RECK 1 3.6926860
Στο παράδειγμα, περάσαμε δύο παραμέτρους Εάν έχει καθοριστεί μόνο η παράμετρος γονιδίων, το αποτέλεσμα είναι μια λίστα επιλεγμένων γονιδίων και όλες οι κατασκευές διεργασίας με τουλάχιστον ένα καθορισμένο γονίδιο.0-1μήτραprocessπαραμέτρους, το αποτέλεσμα είναι ότι όλα τα γονίδια δημιουργούν0-1 Μήτρα γονιδίων που έχουν εκχωρηθεί σε τουλάχιστον μία διεργασία στη λίστα. Λάβετε υπόψη ότι ο καθορισμός μόνο των γονιδίων και των παραμέτρων διεργασίας μπορεί να οδηγήσει σε μια πολύ μεγάλη μήτρα 0-1, με αποτέλεσμα μπερδεμένα αποτελέσματα οπτικοποίησης.
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
-
- # Generate the matrix with a list of selected genes
- chord_genes <- chord_dat(data = circ, genes = EC$genes)
- head(chord_genes)
-
- ## heart development vasculature development blood vessel development
- ## PTK2 0 1 1
- ## GNA13 0 1 1
- ## LEPR 0 1 1
- ## APOE 0 1 1
- ## CXCR4 0 1 1
Αυτό το γράφημα προορίζεται να εμφανίσει ένα μικρότερο υποσύνολο δεδομένων υψηλών διαστάσεων. Υπάρχουν κυρίως δύο παράμετροι που μπορούν να προσαρμοστούν:gene.orderκαιnlfc . Η παράμετρος γονιδίων μπορεί να οριστεί ως 'logFC', 'αλφαβητική', 'κανένα'. Στην πραγματικότητα, γενικά καθορίζουμε την παράμετρο γονιδίων ως logFC, η παράμετρος nlfc είναι μια από τις πιο σημαντικές παραμέτρους αυτής της συνάρτησης, επειδή μπορεί να χειριστεί τον τρόπο με τον οποίο κάθε γονίδιο έχει 0 ή περισσότερες τιμές logFC που παρουσιάζονται στη μήτρα. Επομένως, θα πρέπει να καθορίσουμε παραμέτρους για την αποφυγή σφαλμάτων.
Για παράδειγμα, εάν έχετε έναν πίνακα χωρίς τιμές logFC, πρέπει να ορίσετεnlfc=0 Ή πραγματοποιήστε ανάλυση διαφορικής έκφρασης σε γονίδια κάτω από πολλαπλές συνθήκες ή παρτίδες. Η προεπιλεγμένη τιμή είναι "1" επειδή πιστεύεται ότι τις περισσότερες φορές θα υπάρχει μόνο μία τιμή logFC ανά γονίδιο. Χρησιμοποιήστε την παράμετρο space για να ορίσετε το διάστημα μεταξύ έγχρωμων ορθογωνίων που αντιπροσωπεύουν το logFC. Η παράμετρος gene.size καθορίζει το μέγεθος γραμματοσειράς του ονόματος γονιδίου και το gene.space καθορίζει το μέγεθος του χώρου μεταξύ των ονομάτων γονιδίων.
- chord <- chord_dat(data = circ, genes = EC$genes, process = EC$process)
- GOChord(chord, space = 0.02, gene.order = 'logFC', gene.space = 0.25, gene.size = 5)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
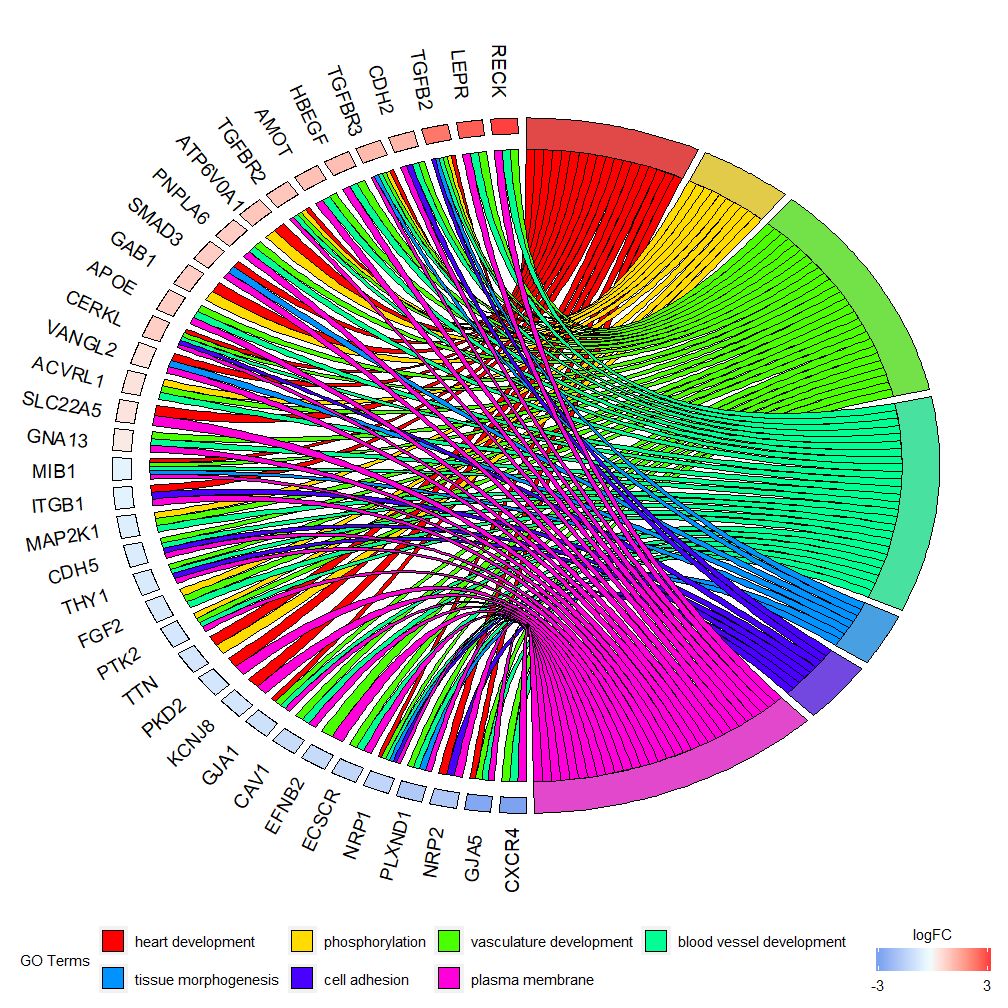
Μπορεί να ρυθμιστεί σύμφωνα με την τιμή logFCgene.order=‘logFC’ , τα γονίδια ταξινομούνται σύμφωνα με τις τιμές logFC τους. Μερικές φορές η εικόνα μπορεί να γίνει λίγο γεμάτη και αυτό μπορεί να αυτοματοποιηθεί χρησιμοποιώντας την παράμετρο ορίου για τη μείωση του αριθμού των γονιδίων ή των οδών που εμφανίζονται. Το όριο είναι ένα διάνυσμα με δύο τιμές αποκοπής (η προεπιλογή είναι c(0,0)). Η πρώτη τιμή καθορίζει τον ελάχιστο αριθμό οδών στις οποίες πρέπει να εκχωρηθεί το γονίδιο. Η δεύτερη τιμή καθορίζει τον αριθμό των γονιδίων που έχουν εκχωρηθεί στο μονοπάτι.
- # 仅显示分配给至少三个通路的基因
- GOChord(chord, limit = c(3, 0), gene.order = 'logFC')
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
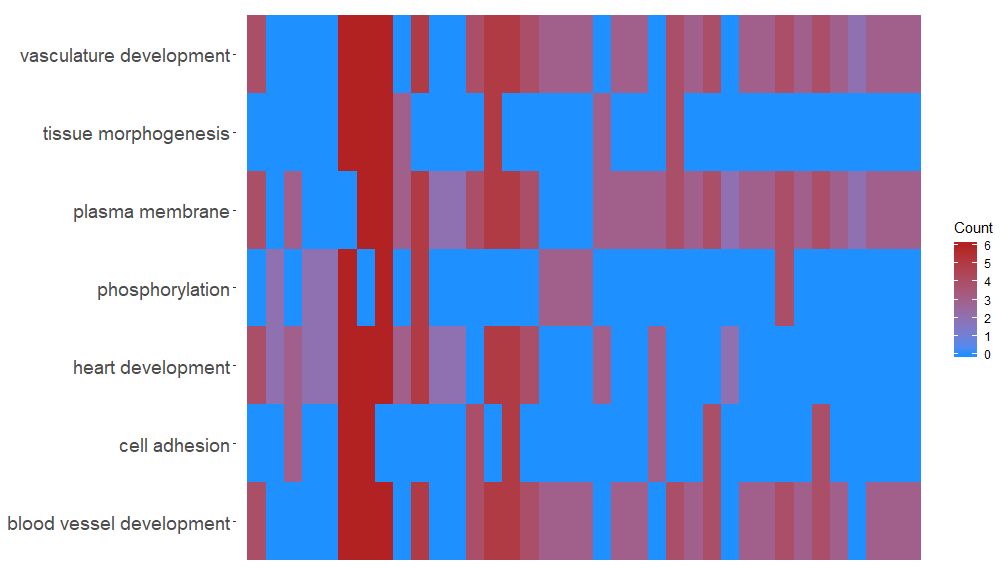
Η συνάρτηση GOHeat μπορεί να εμφανίσει τη σχέση μεταξύ γονιδίων και μονοπατιών χρησιμοποιώντας έναν χάρτη θερμότητας, παρόμοιο με το GOChord. Οι βιολογικές διεργασίες εμφανίζονται οριζόντια και τα γονίδια εμφανίζονται κάθετα. Κάθε στήλη χωρίζεται σε μικρά ορθογώνια και το χρώμα εξαρτάται γενικά από την τιμή logFC. Επιπλέον, συγκεντρώθηκαν γονίδια εμπλουτισμένα σε παρόμοια λειτουργικά μονοπάτια. Υπάρχουν δύο λειτουργίες για την επιλογή χρώματος του heatmap, ανάλογα με τις παραμέτρους nlfc. Εάν nlfc = 0, το χρώμα είναι ο αριθμός των εμπλουτισμένων μονοπατιών για κάθε γονίδιο. Δείτε παραδείγματα για λεπτομέρειες:
- # First, we use the chord object without logFC column to create the heatmap
- GOHeat(chord[,-8], nlfc = 0)
GOHeat(chord[,-8])Το χρώμα αντιστοιχεί στο logFC του γονιδίου στην περίπτωση nlfc = 1
GOHeat(chord, nlfc = 1, fill.col = c('red', 'yellow', 'green'))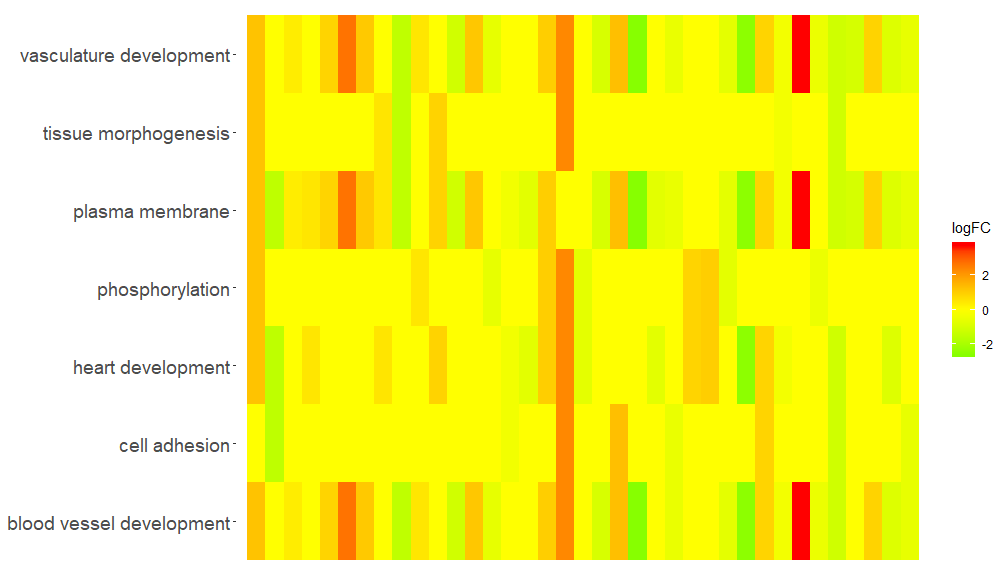
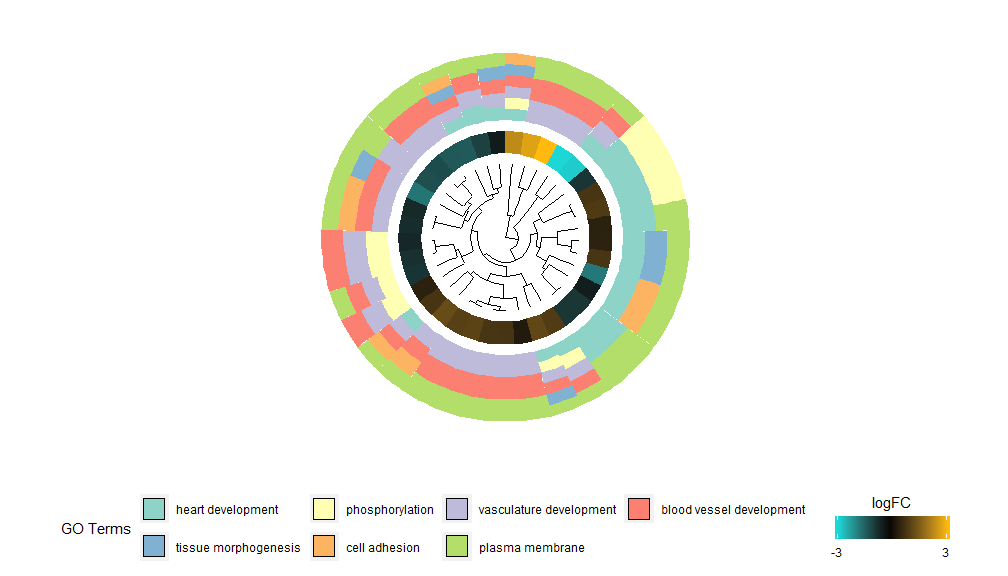
Η ιδέα πίσω από τη λειτουργία GOCluster είναι να εμφανίζει όσο το δυνατόν περισσότερες πληροφορίες. Εδώ είναι ένα παράδειγμα:
- GOCluster(circ, EC$process, clust.by = 'logFC', term.width = 2)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
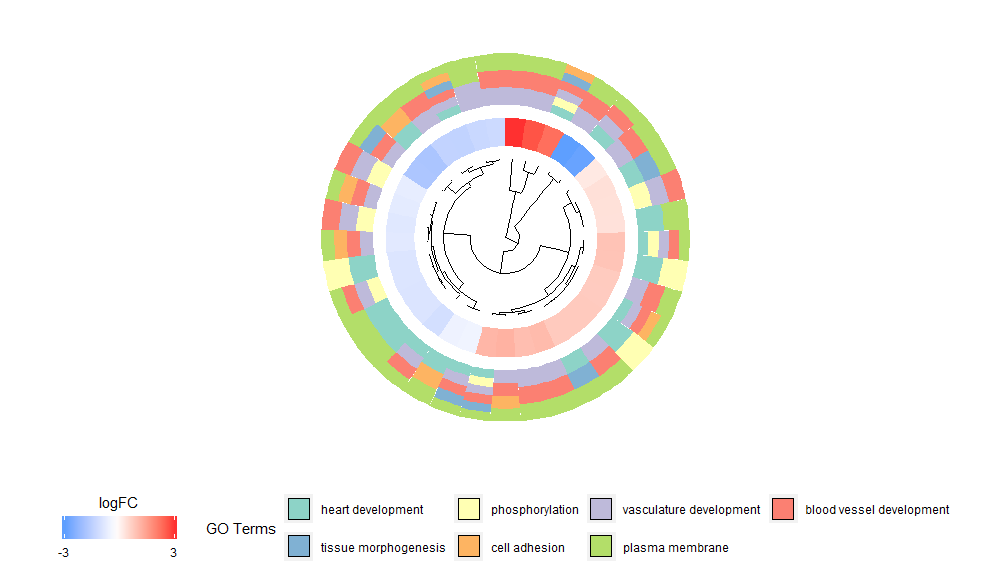
Η ιεραρχική ομαδοποίηση είναι μια δημοφιλής μέθοδος ανάλυσης ομαδοποίησης χωρίς επίβλεψη για γονιδιακή έκφραση που εξασφαλίζει μια αμερόληπτη ομαδοποίηση γονιδίων μεταξύ τους κατά μοτίβο έκφρασης, έτσι ώστε οι συστάδες να μπορούν να περιέχουν πολλαπλές ομάδες συν-ρυθμιζόμενων ή λειτουργικά σχετιζόμενων γονιδίων. Το GOCluster χρησιμοποιεί τοhclust Η μέθοδος εκτελεί ιεραρχική ομαδοποίηση προφίλ γονιδιακής έκφρασης. Εάν θέλετε να αλλάξετε τη μέτρηση απόστασης ή τον αλγόριθμο ομαδοποίησης, χρησιμοποιήστε τις παραμέτρους μέτρηση και συστάδα αντίστοιχα. Το δενδρόγραμμα που προκύπτει μπορεί να μετατραπεί με τη βοήθεια του ggdendro και να απεικονιστεί με το ggplot2. Επιλέξτε μια κυκλική διάταξη, καθώς δεν είναι μόνο αποτελεσματική αλλά και οπτικά ελκυστική. Ο πρώτος κύκλος δίπλα στο δενδρόγραμμα αντιπροσωπεύει το logFC του γονιδίου, το οποίο είναι στην πραγματικότητα το φύλλο του δέντρου ομαδοποίησης. Εάν ενδιαφέρεστε για πολλαπλές αντιθέσεις, μπορείτε να τροποποιήσετε την παράμετρο nlfc, από προεπιλογή έχει οριστεί σε "1", οπότε σχεδιάζεται μόνο ένας δακτύλιος. Οι τιμές logFC κωδικοποιούνται με χρώμα χρησιμοποιώντας μια κλίμακα χρώματος που ορίζεται από το χρήστη (lfc.col), ο επόμενος κύκλος αντιπροσωπεύει τη διαδρομή που έχει εκχωρηθεί στο γονίδιο. Για καλύτερη εμφάνιση, ο αριθμός των καναλιών έχει μειωθεί και το χρώμα των καναλιών μπορεί να αλλάξει χρησιμοποιώντας την παράμετρο term.col.ακόμη διαθέσιμο?GOCluster για να δείτε πώς μπορείτε να αλλάξετε τις παραμέτρους. Η πιο σημαντική παράμετρος αυτής της συνάρτησης είναι το cluster.by, το οποίο μπορεί να καθοριστεί για ομαδοποίηση βάσει μοτίβων έκφρασης γονιδίων («logFC», όπως φαίνεται παραπάνω) ή λειτουργικές κατηγορίες («όροι»).
- GOCluster(circ, EC$process, clust.by = 'term', lfc.col = c('darkgoldenrod1', 'black', 'cyan1'))
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
Τα διαγράμματα Venn μπορούν να χρησιμοποιηθούν για την ανίχνευση σχέσεων μεταξύ διαφόρων καταλόγων διαφορικά εκφραζόμενων γονιδίων ή για τη διερεύνηση της τομής γονιδίων πολλαπλών μονοπατιών σε λειτουργικές αναλύσεις. Τα διαγράμματα Venn δεν δείχνουν μόνο τον αριθμό των επικαλυπτόμενων γονιδίων, αλλά και πληροφορίες σχετικά με το πρότυπο έκφρασης του γονιδίου (συνήθως ρυθμίζεται προς τα πάνω, συχνά ρυθμίζεται προς τα κάτω ή αντίθετα ρυθμίζεται). Επί του παρόντος, έως και τρία σύνολα δεδομένων χρησιμοποιούνται ως είσοδοι. Τα δεδομένα εισόδου περιέχουν τουλάχιστον δύο στήλες: μία για ονόματα γονιδίων και μία για τιμές logFC.
- l1 <- subset(circ, term == 'heart development', c(genes,logFC))
- l2 <- subset(circ, term == 'plasma membrane', c(genes,logFC))
- l3 <- subset(circ, term == 'tissue morphogenesis', c(genes,logFC))
- GOVenn(l1,l2,l3, label = c('heart development', 'plasma membrane', 'tissue morphogenesis'))
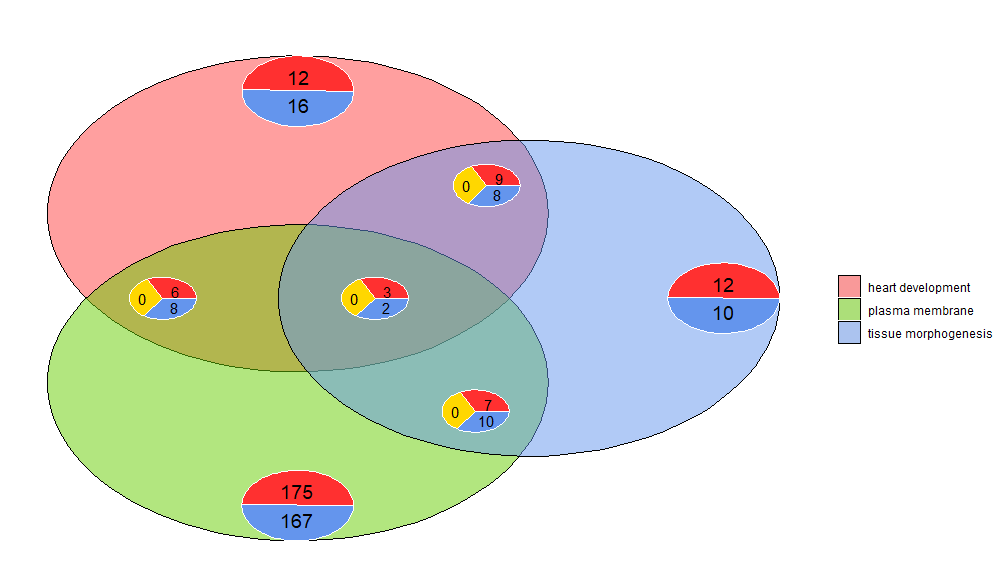
Για παράδειγμα, η καρδιακή ανάπτυξη και η μορφογένεση των ιστών έχουν 22 γονίδια, 12 ρυθμίζονται προς τα πάνω και 10 ρυθμίζονται προς τα κάτω. Ένα σημαντικό πράγμα που πρέπει να σημειωθεί είναι ότι τα γραφήματα πίτας δεν εμφανίζουν περιττές πληροφορίες. Επομένως, εάν συγκριθούν τρία σύνολα δεδομένων, τα γονίδια κοινά σε όλα τα σύνολα δεδομένων (το διάγραμμα μεσαίας πίτας) δεν περιλαμβάνονται στα άλλα γραφήματα πίτας. Αυτό το εργαλείο είναι διαθέσιμο στο shinyapp https://wwalter.shinyapps.io/Venn/, το εργαλείο web είναι πιο διαδραστικό, ο κύκλος έχει μια περιοχή ανάλογη με τον αριθμό των γονιδίων στο σύνολο δεδομένων και το ρυθμιστικό μπορεί να χρησιμοποιηθεί για τη μετακίνηση του μικρό διάγραμμα πίτας και διαθέτει GOVenn Διαθέτει όλες τις επιλογές για αλλαγή της διάταξης της πλοκής και επίσης λήψη εικόνων και λιστών γονιδίων.
Αρχική σελίδα λογισμικού: https://wencke.github.io/

Έχει αφοσιωθεί στην έρευνα της τεχνολογίας για περισσότερα από τριάντα χρόνια και είναι ικανός σε διάφορες γλώσσες όπως java, linux, javascript, php, css, κ.λπ., και έχει κάνει πολλές συνεισφορές στον τομέα του ανοιχτού κώδικα ένας σταθμός τεκμηρίωσης προγραμματιστή για κοινή χρήση ορισμένων ζητημάτων στην ανάπτυξη τεχνολογίας για μελλοντική αναφορά

Ταχυδρομείο[email protected]