
minhas informações de contato
Correspondência[email protected]
2024-07-12
한어Русский языкEnglishFrançaisIndonesianSanskrit日本語DeutschPortuguêsΕλληνικάespañolItalianoSuomalainenLatina
O pacote GOPlot é utilizado para visualização de dados biológicos. Em vez disso, este pacote integra e visualiza dados de expressão com os resultados de análises funcionais.Mas tenha cuidadoEste pacote não pode ser utilizado para realizar essas análises, apenas para visualizar os resultados. . Em todos os campos da ciência, é difícil descrever as coisas de forma realista devido às restrições de espaço e à simplicidade necessária para os resultados, pelo que a informação precisa de ser visualizada e as imagens utilizadas para transmitir a informação. Gráficos bem projetados fornecem mais informações em menos espaço. A ideia do pacote é permitir que os usuários examinem rapidamente grandes quantidades de dados, revelem tendências nos dados e encontrem padrões e correlações nos dados.
A visualização de dados pode nos ajudar a encontrar respostas para questões biológicas, julgar uma determinada hipótese e até descobrir diferentes ângulos para investigar diferentes problemas. E as funções de plotagem deste pacote são desenvolvidas com base na estrutura hierárquica dos dados, começando com os dados gerais e terminando com um subconjunto de genes selecionados e caminhos correspondentes.
Vamos explicar concretamente com um exemplo.
Chamamos os dados que vêm com GOplot, que vem do GEOGSE47067, contendo informações do transcriptoma de células endoteliais de dois tecidos (cérebro e coração). Para obter mais informações, consulte o artigo de Nolan et al.Os dados são normalizados e genes expressos diferencialmente são encontrados.e, em seguida, use a ferramenta de anotação da função DAVID (os dados de anotação DAVID são atualizados lentamente e não são recomendados agora. Recomenda-se usarGo East, a melhor ferramenta online de análise de enriquecimento GOeEste site, que pode realizar análises de enriquecimento em apenas uma etapa, foi citado mais de 350 vezes pelo CNS e outros antes de ser publicado.Realizar análise de enriquecimento,Domine GSEA em um artigo, tutorial super detalhado) Anotação genética de genes expressos diferencialmente (adjusted p-value < 0.05 ) e análise de enriquecimento funcional. Este conjunto de dados contém as seguintes cinco categorias de dados:
| nome | descrever | Tamanho do conjunto de dados |
|---|---|---|
| EC$eset | Expressão gênica normalizada em células endoteliais do cérebro e do coração (3 repetições) | 20644 x 7 |
| EC$genelista | Genes expressos diferencialmente (valor p ajustado < 0,05) | 2039 x 7 |
| EC$david | Resultados da análise de enriquecimento funcional de genes diferenciais usando DAVID | 174 x 5 |
| EC$gene | Genes e logFC | 37 x 2 |
| Processo EC$ | Vetores de recursos selecionados para processos biológicos enriquecidos | 7 |
Queremos ver as vias enriquecidas por GO de genes expressos diferencialmente, mas antes de começarmos a desenhar, precisamos fornecer dados que atendam aos requisitos de formato.De modo geral, os dados necessários para desenhar o gráfico são fornecidos por você, masExiste uma função neste pacotecircle_datPode nos ajudar a lidar com o formato dos dados。circle_datPode combinar os resultados da análise de enriquecimento funcional de genes selecionados e seus valores logFC, principalmente para genes expressos diferencialmente.circle_dat O uso é muito simples, basta ler dois dados. Os primeiros dados contêm os resultados da análise de enriquecimento funcional, com pelo menos quatro colunas (categoria de análise de enriquecimento funcional, via, gene, valor p ajustado).Os segundos dados são do gene selecionado e seu logFC, esses dados podem ser a fontelimmaOs resultados da análise estatística (Nota das biografias: certifique-se de prestar atenção a dois arquivosComo os genes são nomeadosSeja consistente, como todosGene symbol ). Vejamos os formatos de dados mencionados acima com exemplos.
- #安装已发布的稳定版本
- #install.packages('GOplot')
- #安装github上的开发版本
- #install_github('wencke/wencke.github.io')
- #载入包
- library(GOplot)
- #读入包内自带的数据
- data(EC)
- #查看功能富集分析结果的数据格式
- head(EC$david)
-
- ## Category ID Term
- ## 1 BP GO:0007507 heart development
- ## 2 BP GO:0001944 vasculature development
- ## 3 BP GO:0001568 blood vessel development
- ## 4 BP GO:0048729 tissue morphogenesis
- ## 5 BP GO:0048514 blood vessel morphogenesis
- ## 6 BP GO:0051336 regulation of hydrolase activity
- ## Genes
- ## 1 DLC1, NRP2, NRP1, EDN1, PDLIM3, GJA1, TTN, GJA5, ZIC3, TGFB2, CERKL, GATA6, COL4A3BP, GAB1, SEMA3C, MKL2, SLC22A5, MB, PTPRJ, RXRA, VANGL2, MYH6, TNNT2, HHEX, MURC, MIB1, FOXC2, FOXC1, ADAM19, MYL2, TCAP, EGLN1, SOX9, ITGB1, CHD7, HEXIM1, PKD2, NFATC4, PCSK5, ACTC1, TGFBR2, NF1, HSPG2, SMAD3, TBX1, TNNI3, CSRP3, FOXP1, KCNJ8, PLN, TSC2, ATP6V0A1, TGFBR3, HDAC9
- ## 2 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, EFNB2, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 3 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 4 DLC1, ENAH, NRP1, PGF, ZIC2, TGFB2, CD44, ILK, SEMA3C, RET, AR, RXRA, VANGL2, LEF1, TNNT2, HHEX, MIB1, NCOA3, FOXC2, FOXC1, TGFB1I1, WNT5A, COBL, BBS4, FGFR3, TNC, BMPR2, CTNND1, EGLN1, NR3C1, SOX9, TCF7L1, IGF1R, FOXQ1, MACF1, HOXA5, BCL2, PLXND1, CAR2, ACTC1, TBX4, SMAD3, FZD3, SHANK3, FZD6, HOXB4, FREM2, TSC2, ZIC5, TGFBR3, APAF1
- ## 5 GNA13, CAV1, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, CDH2, MEIS1, WT1, TGFB2, WARS, PTK2, CERKL, APOE, CXCR4, ANG, SEMA3C, PLCD1, NOS2, MKL2, PLXND1, FIGF, FGF2, PTPRJ, TGFBR2, TBX4, NF1, TBX1, TNNI3, PNPLA6, VASH1, THY1, NUS1, MEOX2, ECSCR, AMOT, HBEGF, FOXC2, FOXC1, HDAC7
- ## 6 CAV1, XIAP, AGFG1, ADORA2A, TNNC1, TBC1D9, LEPR, ABHD5, EDN1, ASAP2, ASAP3, SMAP1, TBC1D12, ANG, TBC1D14, MTCH1, TBC1D13, TBC1D4, TBC1D30, DHCR24, HIP1, VAV3, NOS1, NF1, MYH6, RICTOR, TBC1D22A, THY1, PLCE1, RNF7, NDEL1, CHML, IFT57, ACAP2, TSC2, ERN1, APAF1, ARAP3, ARAP2, ARAP1, HTR2A, F2R
- ## adj_pval
- ## 1 0.000002170
- ## 2 0.000010400
- ## 3 0.000007620
- ## 4 0.000119000
- ## 5 0.000720000
- ## 6 0.001171166
-
- #查看基因的数据格式
- head(EC$genelist)
-
- ## ID logFC AveExpr t P.Value adj.P.Val B
- ## 1 Slco1a4 6.645388 1.2168670 88.65515 1.32e-18 2.73e-14 29.02715
- ## 2 Slc19a3 6.281525 1.1600468 69.95094 2.41e-17 2.49e-13 27.62917
- ## 3 Ddc 4.483338 0.8365231 65.57836 5.31e-17 3.65e-13 27.18476
- ## 4 Slco1c1 6.469384 1.3558865 59.87613 1.62e-16 8.34e-13 26.51242
- ## 5 Sema3c 5.515630 2.3252117 58.53141 2.14e-16 8.81e-13 26.33626
- ## 6 Slc38a3 4.761755 0.9218670 54.11559 5.58e-16 1.76e-12 25.70308
Depois de compreender os dois formatos de dados de entrada, você pode usarcirlce_datfunção para gerar dados de desenho.
- # 生成画图所需的数据格式
- circ <- circle_dat(EC$david, EC$genelist)
-
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
circO objeto possui oito colunas de dados, a saber
categoria: BP (Processo Biológico), CC (Componente Celular) ou MF (Função Molecular)
ID: GO id (coluna opcional, se você quiser usar uma ferramenta de análise funcional que não seja baseada no GO id, você não pode selecionar a coluna ID; o ID aqui também pode ser KEGG ID)
termo: caminho GO
contagem: número de genes em cada via
gene: nome do gene - logFC: valor logFC de cada gene
adj_pval: valor de p ajustado, caminhos com adj_pval<0,05 são considerados significativamente enriquecidos
zscore: zscore não se refere a um método de normalização estatística, mas é um valor facilmente calculado para estimar se um processo biológico (/função molecular/componente celular) tem maior probabilidade de diminuir (valor negativo) ou aumentar (valor positivo).O método de cálculo é o número de genes regulados positivamente menos o número de genes regulados negativamente dividido pela raiz quadrada do número de genes em cada via.
Quando olhamos os dados pela primeira vez, queremos mostrar o maior número possível de caminhos no gráfico e também queremos encontrar caminhos valiosos, por isso precisamos de alguns parâmetros para avaliar a importância. Os gráficos de barras são frequentemente usados para descrever dados de amostra, portanto, podemos usar a função GOBar para criar rapidamente um gráfico de barras bonito.
Primeiro, um gráfico de barras simples é gerado diretamente. O eixo horizontal é gerado.GO Terms, de acordo com seuszscoreClassifique as barras;-log(adj p-value);A cor representazscore, azul indicaz-scorefor um valor negativo, é mais provável que a expressão gênica na via correspondente diminua, indicada em vermelhoz-score for um valor positivo, é mais provável que a expressão genética na via correspondente aumente. Se desejar, a ordem pode ser alterada definindo o parâmetro order.by.zscore como FALSE, caso em que as barras são ordenadas com base em seu significado.
- # 生成简单的条形图
- GOBar(subset(circ, category == 'BP'))
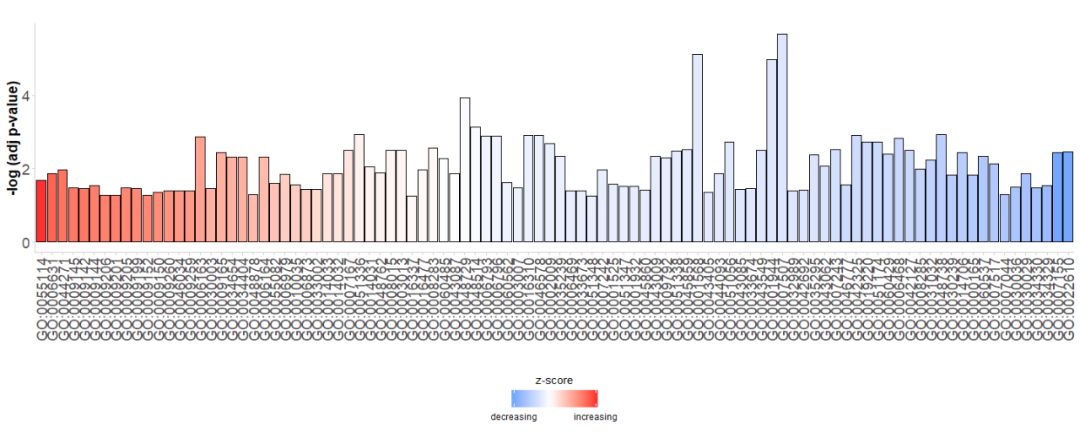
#GOBar(subset(circ, category == 'BP',order.by.zscore=FALSE))Além disso, altere o parâmetro de exibição para desenhar um gráfico de barras de acordo com a categoria do canal.
- #根据通路的类别来绘制条形图
- GOBar(circ, display = 'multiple')
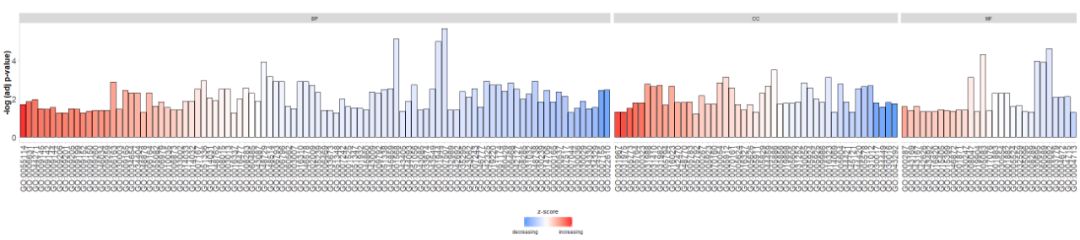
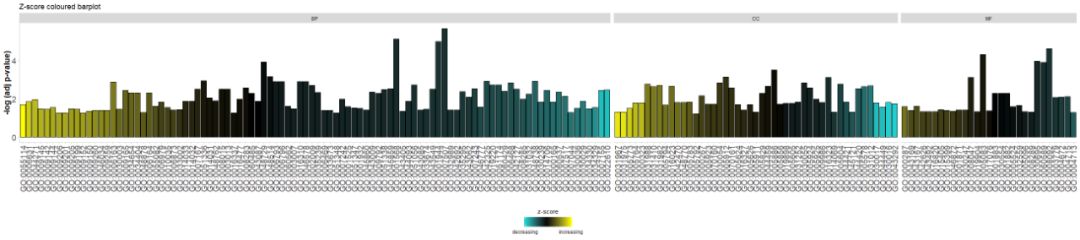
Adicione um título e use parâmetroszsc.colMudarzscoreé cor.
- # Facet the barplot, add a title and change the colour scale for the z-score
- GOBar(circ, display = 'multiple', title = 'Z-score coloured barplot', zsc.col = c('yellow', 'black', 'cyan'))
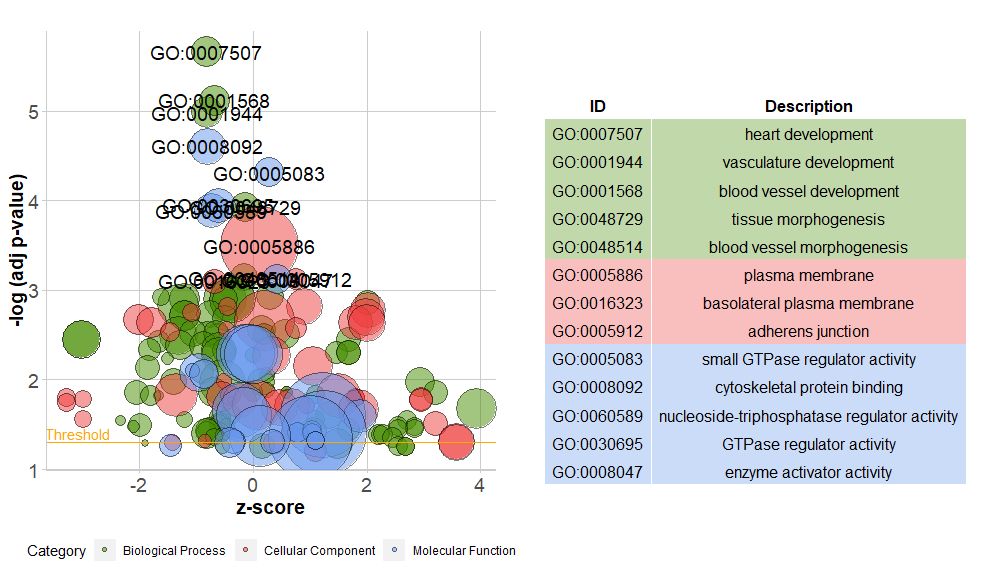
Os gráficos de barras são muito comuns e fáceis de entender, mas podemos usar gráficos de bolhas para exibir mais informações sobre os dados.
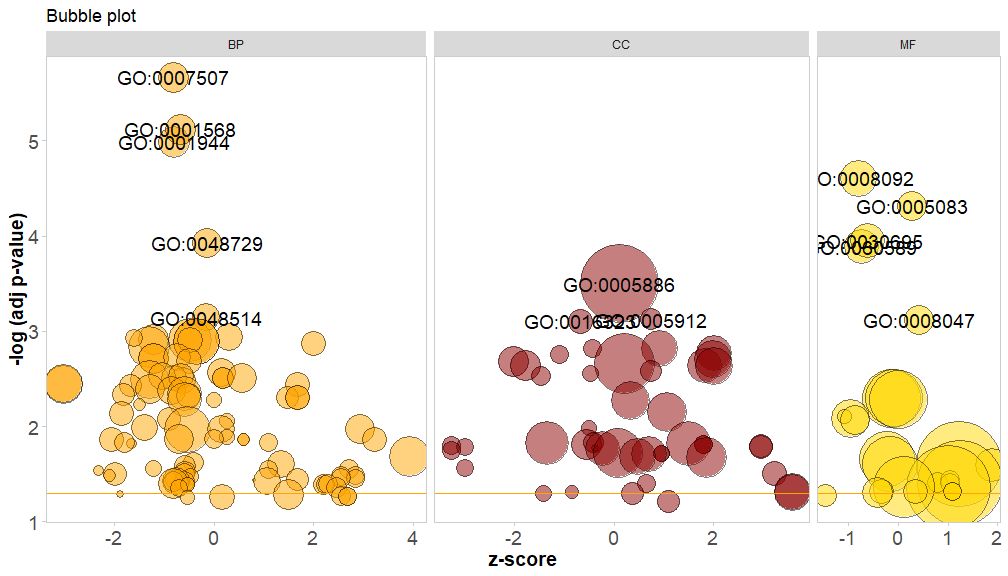
O eixo horizontal ézscore;O eixo vertical é-log(adj p-value), semelhante a um gráfico de barras, quanto maior for, mais significativo será o enriquecimento da área do círculo em relação ao número de genes na via correspondente (;circ$count ); a cor corresponde à categoria correspondente à via, o verde é o processo biológico, o vermelho é o componente celular e o azul é a função molecular.Pode ser inserido por?GOBubble Consulte a página de ajuda da função GOBubble para alterar todos os parâmetros da imagem. Por padrão, cada círculo é marcado com um GO ID correspondente, e uma tabela mostrando a relação correspondente entre GO ID e termo GO também é exibida à direita.Os parâmetros podem ser definidos portable.legendparaFALSE para escondê-lo. Se desejar exibir a descrição do caminho, defina o ID do parâmetro como FALSE.No entanto, devido ao espaço limitado e aos círculos sobrepostos, nem todos os círculos estão marcados, apenas os-log(adj p-value) > 3(o padrão é 5).
- # 生成泡泡图,并展示-log(adj p-value) > 3 的通路的GO ID
- GOBubble(circ, labels = 3)
Se desejar adicionar um título ao gráfico de bolhas ou especificar a cor do círculo e exibir os caminhos de cada categoria separadamente e alterar o limite de GO ID exibido, você poderá adicionar os seguintes parâmetros:
GOBubble(circ, title = 'Bubble plot', colour = c('orange', 'darkred', 'gold'), display = 'multiple', labels = 3)
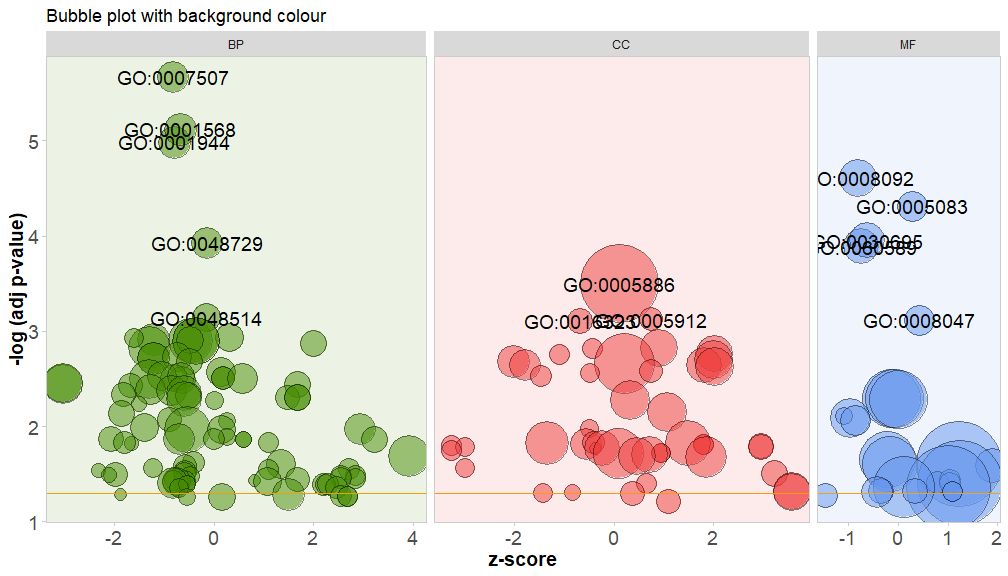
Pinte o fundo da classe do canal definindo o parâmetro bg.col como TRUE.
GOBubble(circ, title = 'Bubble plot with background colour', display = 'multiple', bg.col = T, labels = 3)
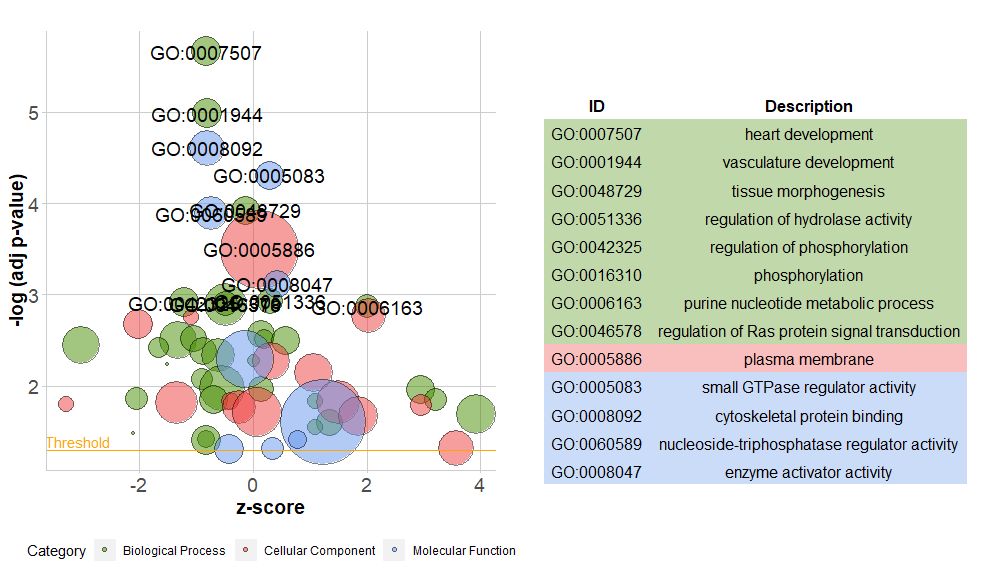
A nova versão do pacote contém uma nova funçãoreduce_overlap , esta função pode reduzir o número de itens redundantes, ou seja, pode excluir todas as vias cuja sobreposição genética é maior ou igual ao limite definido, e reter apenas uma via de cada grupo como representativa, independentemente da exibição de todos caminhos em GO. Ao reduzir o número de termos redundantes, a legibilidade dos gráficos (como gráficos de bolhas) é significativamente melhorada.
- # reduce_overlap,参数设置为0.75
- reduced_circ <- reduce_overlap(circ, overlap = 0.75)
-
- GOBubble(reduced_circ, labels = 2.8)
Embora um gráfico que mostre todas as informações possa nos ajudar a descobrir quais caminhos são mais significativos, a realidade ainda depende das hipóteses e ideias que você deseja confirmar com os dados, e os caminhos mais importantes podem não ser necessariamente aqueles nos quais você está interessado. Portanto, ao selecionar manualmente um conjunto valioso de caminhos (EC$process ), precisamos de um diagrama que nos mostre informações mais detalhadas sobre esse conjunto específico de caminhos.Mas surge um problema na apresentação destes números: por vezes é difícil interpretarzscore Informação fornecida.Afinal, este método de cálculo não é universal Como mostrado acima, é simplesmente o número de genes regulados positivamente menos o número de genes regulados negativamente dividido pela raiz quadrada do número de genes em cada via, usando.GOCircleO gráfico resultante também enfatiza esse fato.
O círculo externo do diagrama circular mostra o valor logFC dos genes de cada via como pontos dispersos. Os círculos vermelhos indicam regulação positiva e os azuis indicam regulação negativa.Parâmetros podem ser usadoslfc.col Mudar cor. Isto também explica porque, em alguns casos, caminhos muito importantes têm zscores próximos de zero. Um zscore igual a zero não significa que o canal não seja importante. Isso apenas mostra que o zscore é uma medida aproximada, porque obviamente o zscore também não leva em consideração o nível funcional e a dependência de ativação de genes individuais em processos biológicos.
GOCircle(circ)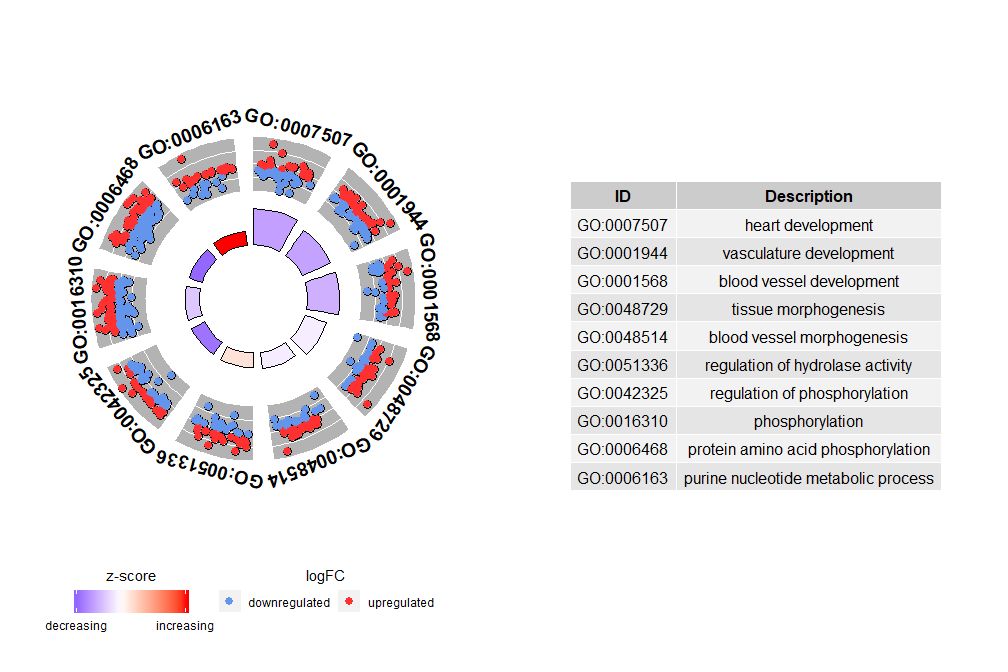
nsub Os parâmetros podem ser números definidos ou vetores de caracteres. Se for um vetor de caracteres, ele contém o GO ID ou caminho a ser exibido;
- # 生成特定通路的圈图
- IDs <- c('GO:0007507', 'GO:0001568', 'GO:0001944', 'GO:0048729', 'GO:0048514', 'GO:0005886', 'GO:0008092', 'GO:0008047')
- GOCircle(circ, nsub = IDs)
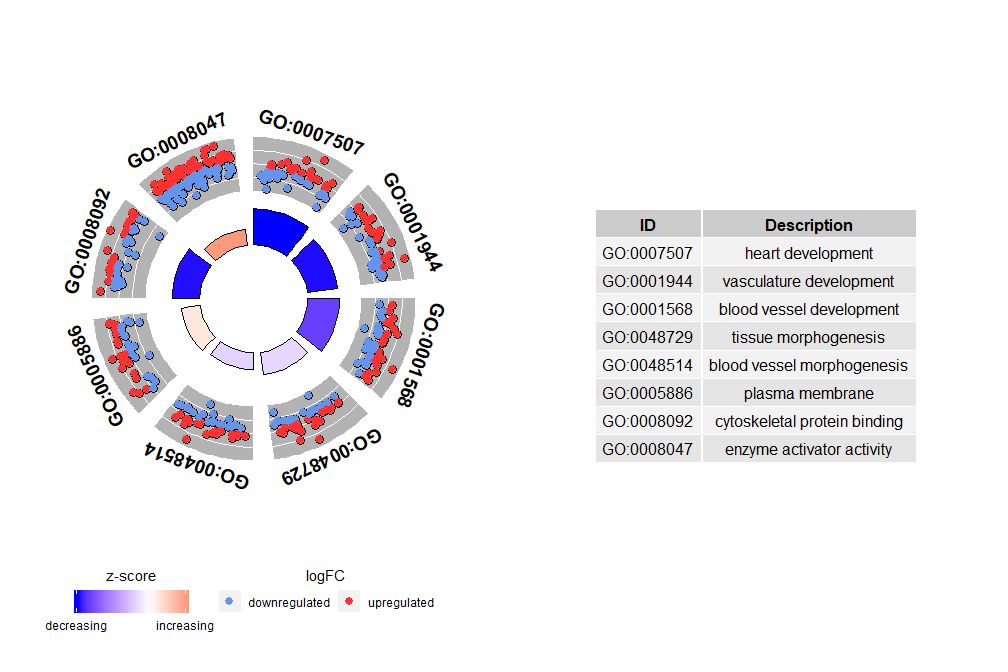
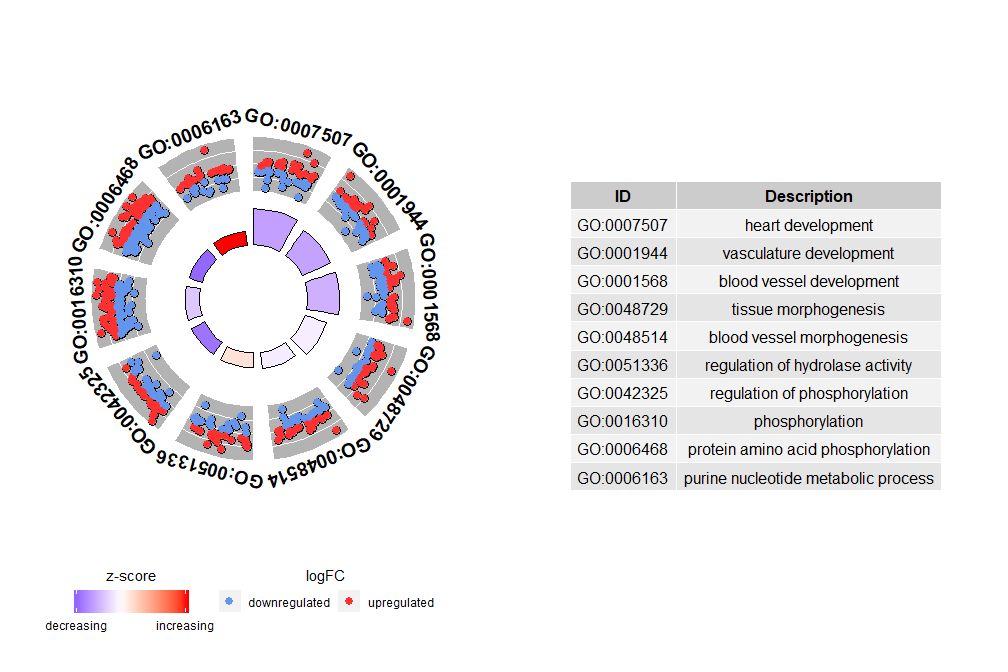
Se nsub for um vetor numérico, o número definirá o número a ser exibido. Começa na primeira linha do dataframe de entrada. Esta visualização só funciona com dados menores. O número máximo de canais é padronizado como 12. Embora o número de canais seja reduzido, a quantidade de informações exibidas aumenta.
- # 圈图展示数据前十个通路
- GOCircle(circ, nsub = 10)
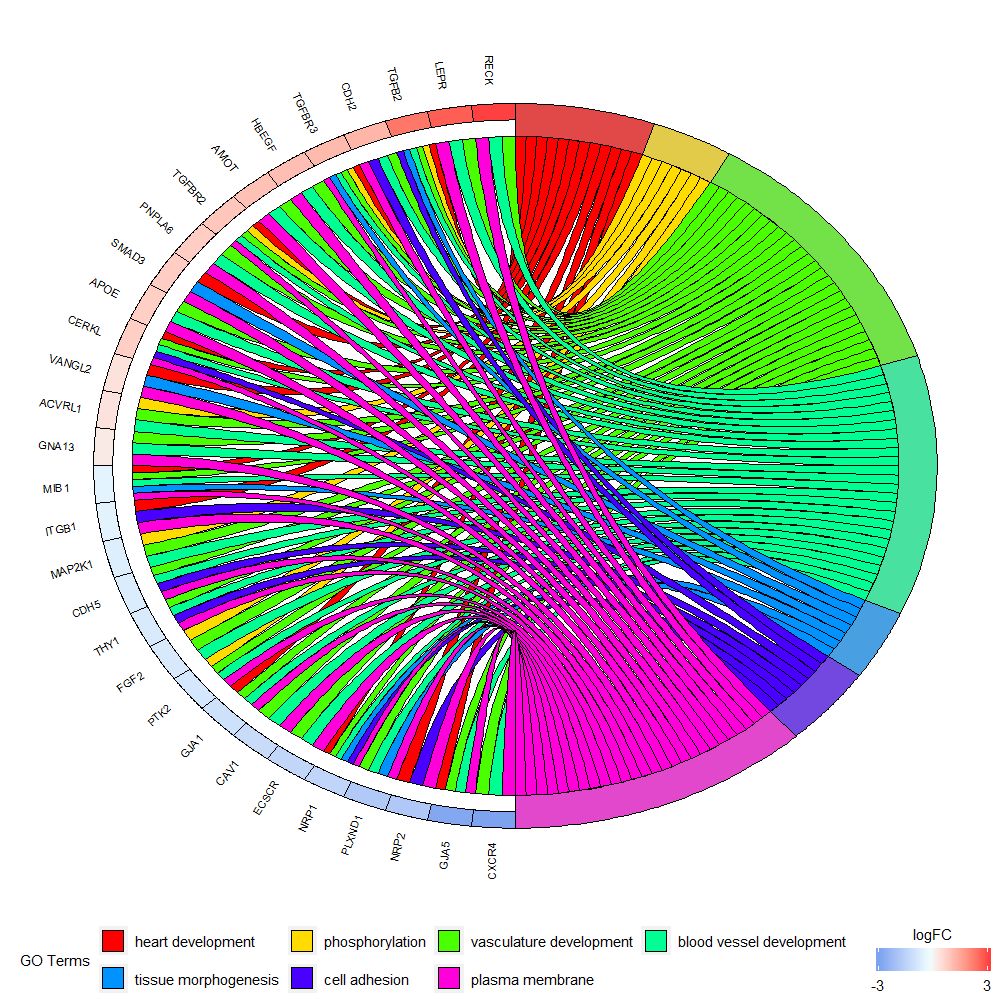
GOChord pode exibir a relação entre genes e caminhos selecionados e o logFC dos genes.Primeiro você precisa inserir uma matriz, que você mesmo pode construir0-1Matrix, você também pode usar funçõeschord_dat Construir. Esta função possui três parâmetros: dados, genes e processo, dos quais os dois últimos parâmetros devem ter pelo menos um parâmetro.Então a funçãocircle_datCombine dados de expressão com resultados de análises funcionais.
Gráficos de barras e gráficos de bolhas podem dar uma primeira impressão dos dados. Agora, você pode selecionar alguns genes e caminhos que consideramos valiosos. Embora o GOCircle adicione uma camada para exibir o valor de expressão dos genes nos caminhos, falta uma única informação. nas relações entre genes e múltiplas vias. Não é fácil descobrir se certos genes estão ligados a múltiplos processos. GOChord compensa as deficiências do GOCircle. As linhas dos dados gerados são genes e as colunas são caminhos. “0” significa que o gene não está atribuído ao caminho e “1” é o oposto.
- # 找到感兴趣的的基因,这里我们以EC$genes为例
- head(EC$genes)
-
- ## ID logFC
- ## 1 PTK2 -0.6527904
- ## 2 GNA13 0.3711599
- ## 3 LEPR 2.6539788
- ## 4 APOE 0.8698346
- ## 5 CXCR4 -2.5647537
- ## 6 RECK 3.6926860
-
- # 获得感兴趣基因的通路
- EC$process
-
- ## [1] "heart development" "phosphorylation"
- ## [3] "vasculature development" "blood vessel development"
- ## [5] "tissue morphogenesis" "cell adhesion"
- ## [7] "plasma membrane"
-
- # 使用chord_dat构建矩阵
- chord <- chord_dat(circ, EC$genes, EC$process)
- head(chord)
-
- ## heart development phosphorylation vasculature development
- ## PTK2 0 1 1
- ## GNA13 0 0 1
- ## LEPR 0 0 1
- ## APOE 0 0 1
- ## CXCR4 0 0 1
- ## RECK 0 0 1
- ## blood vessel development tissue morphogenesis cell adhesion
- ## PTK2 1 0 0
- ## GNA13 1 0 0
- ## LEPR 1 0 0
- ## APOE 1 0 0
- ## CXCR4 1 0 0
- ## RECK 1 0 0
- ## plasma membrane logFC
- ## PTK2 1 -0.6527904
- ## GNA13 1 0.3711599
- ## LEPR 1 2.6539788
- ## APOE 1 0.8698346
- ## CXCR4 1 -2.5647537
- ## RECK 1 3.6926860
No exemplo, passamos dois parâmetros. Se apenas o parâmetro genes for especificado, o resultado será uma lista de genes selecionados e todas as construções de processos com pelo menos um gene especificado.0-1matriz; se apenas especificadoprocessparâmetros, o resultado é que todos os genes geram0-1 Matriz de genes atribuídos a pelo menos um processo da lista. Esteja ciente de que especificar apenas os genes e os parâmetros do processo pode resultar em uma matriz 0-1 muito grande, resultando em resultados de visualização confusos.
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
-
- # Generate the matrix with a list of selected genes
- chord_genes <- chord_dat(data = circ, genes = EC$genes)
- head(chord_genes)
-
- ## heart development vasculature development blood vessel development
- ## PTK2 0 1 1
- ## GNA13 0 1 1
- ## LEPR 0 1 1
- ## APOE 0 1 1
- ## CXCR4 0 1 1
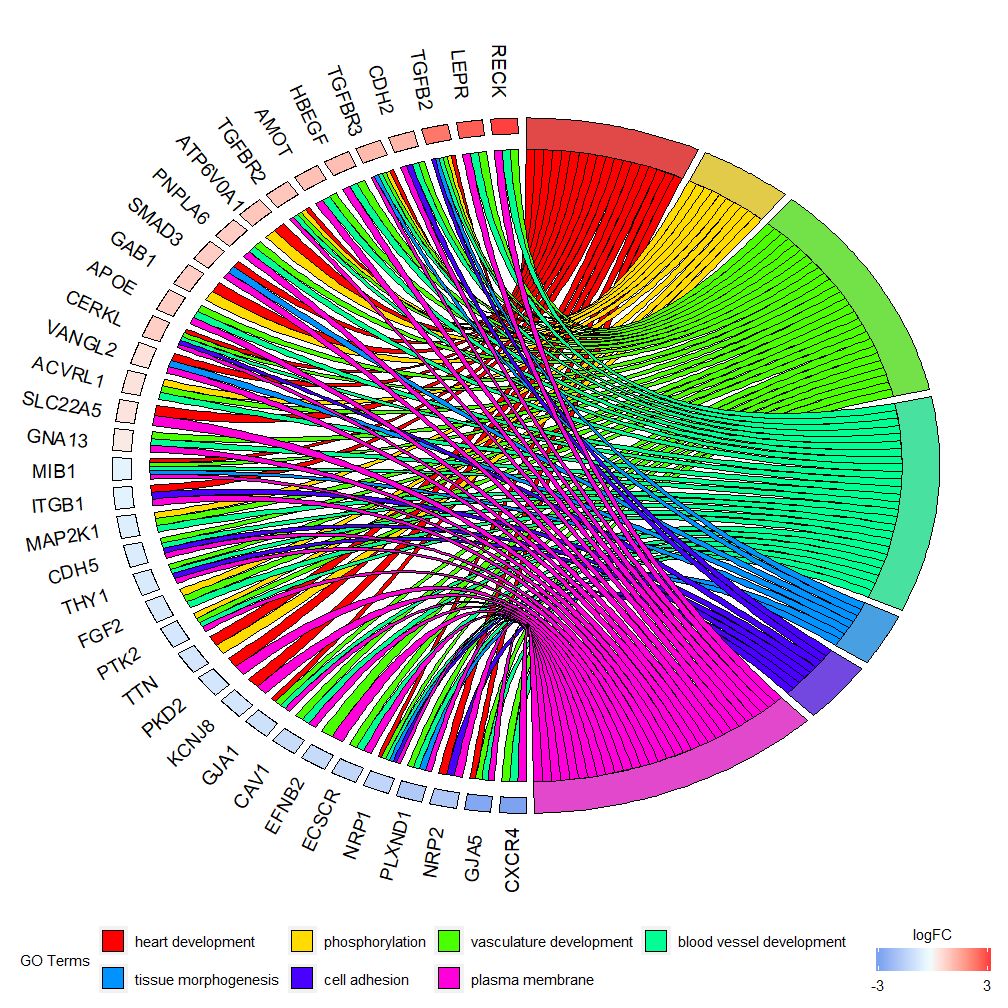
Este gráfico tem como objetivo mostrar um subconjunto menor de dados de alta dimensão. Existem basicamente dois parâmetros que podem ser ajustados:gene.orderenlfc . O parâmetro genes pode ser especificado como 'logFC', 'alfabético', 'none'. Na verdade, geralmente especificamos o parâmetro genes como logFC; o parâmetro nlfc é um dos parâmetros mais importantes desta função, pois pode lidar com como cada gene tem 0 ou mais valores logFC apresentados na matriz. Portanto devemos especificar parâmetros para evitar erros.
Por exemplo, se você tiver uma matriz sem valores logFC, deverá definirnlfc=0 ; Ou execute análise de expressão diferencial em genes sob múltiplas condições ou lotes. Nesse caso, cada gene contém vários valores logFC e o número da coluna nlfc=logFC precisa ser definido. O valor padrão é “1” porque acredita-se que na maioria das vezes haverá apenas um valor logFC por gene. Use o parâmetro space para definir o espaço entre retângulos coloridos que representam logFC. O parâmetro gene.size especifica o tamanho da fonte do nome do gene e gene.space especifica o tamanho do espaço entre os nomes dos genes.
- chord <- chord_dat(data = circ, genes = EC$genes, process = EC$process)
- GOChord(chord, space = 0.02, gene.order = 'logFC', gene.space = 0.25, gene.size = 5)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
Pode ser definido de acordo com o valor logFCgene.order=‘logFC’ , os genes são classificados de acordo com seus valores logFC. Às vezes, a imagem pode ficar um pouco lotada e isso pode ser automatizado usando o parâmetro limit para reduzir o número de genes ou caminhos mostrados. Limite é um vetor com dois valores de corte (o padrão é c(0,0)). O primeiro valor especifica o número mínimo de vias às quais o gene deve ser atribuído. O segundo valor determina o número de genes atribuídos à via.
- # 仅显示分配给至少三个通路的基因
- GOChord(chord, limit = c(3, 0), gene.order = 'logFC')
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
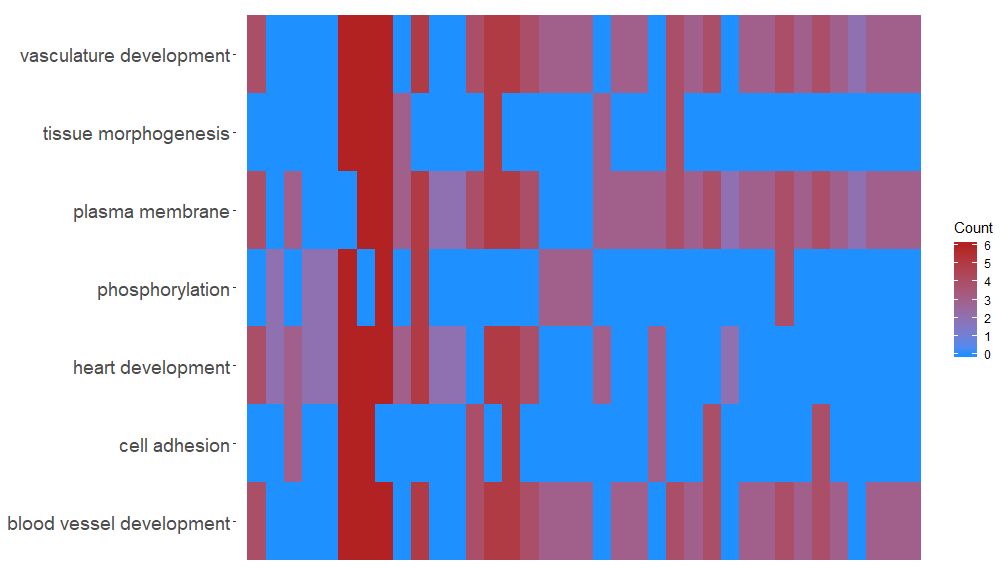
A função GOHeat pode exibir a relação entre genes e caminhos usando um mapa de calor, semelhante ao GOChord. Os processos biológicos são exibidos horizontalmente e os genes são exibidos verticalmente. Cada coluna é dividida em pequenos retângulos e a cor geralmente depende do valor logFC. Além disso, genes enriquecidos em vias funcionais semelhantes foram agrupados. Existem dois modos para seleção de cores do mapa de calor, dependendo dos parâmetros nlfc. Se nlfc = 0, a cor é o número de vias enriquecidas para cada gene. Veja exemplos para detalhes:
- # First, we use the chord object without logFC column to create the heatmap
- GOHeat(chord[,-8], nlfc = 0)
GOHeat(chord[,-8])A cor corresponde ao logFC do gene no caso nlfc = 1
GOHeat(chord, nlfc = 1, fill.col = c('red', 'yellow', 'green'))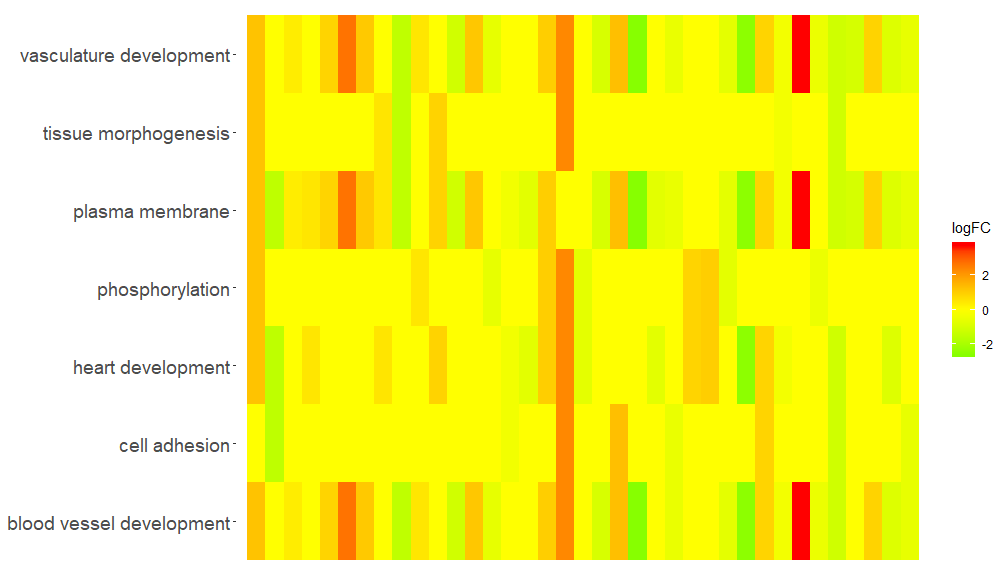
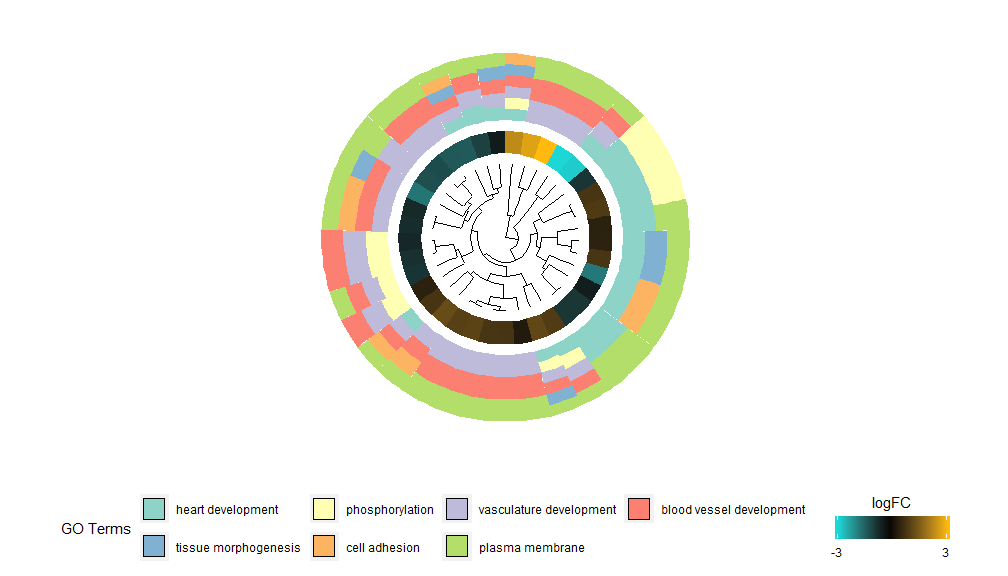
A ideia por trás da funcionalidade GOCluster é exibir o máximo de informações possível. Aqui está um exemplo:
- GOCluster(circ, EC$process, clust.by = 'logFC', term.width = 2)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
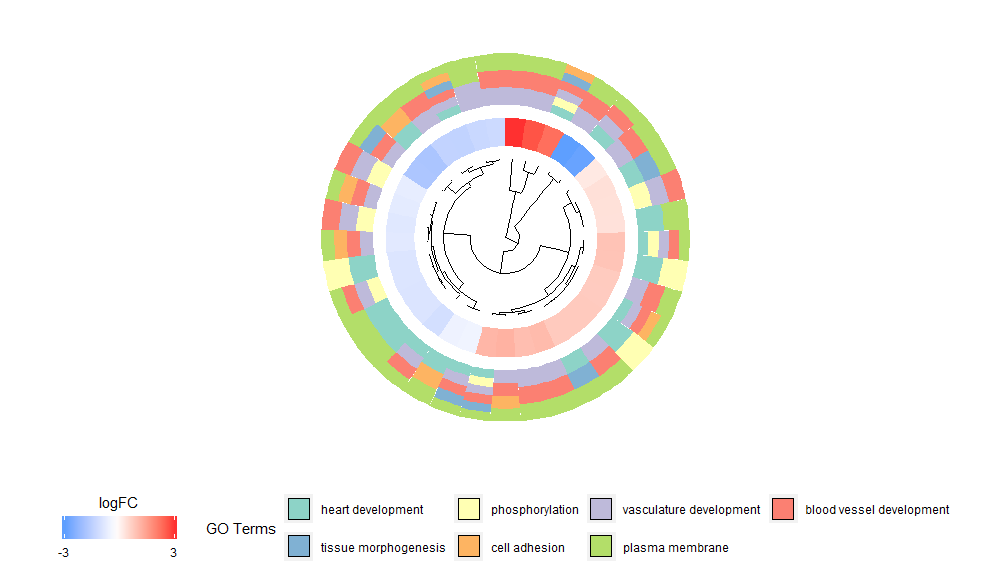
O agrupamento hierárquico é um método popular de análise de agrupamento não supervisionado para expressão gênica que garante um agrupamento imparcial de genes por padrão de expressão, de modo que os agrupamentos possam conter vários grupos de genes co-regulados ou funcionalmente relacionados. GOCluster usa ohclust O método realiza agrupamento hierárquico de perfis de expressão gênica. Se você deseja alterar a métrica de distância ou o algoritmo de cluster, use os parâmetros metric e clust respectivamente. O dendograma resultante pode ser convertido com a ajuda do ggdendro e visualizado com o ggplot2. Escolha um layout circular, pois não é apenas eficaz, mas também visualmente atraente. O primeiro círculo próximo ao dendograma representa o logFC do gene, que na verdade é a folha da árvore de agrupamento. Se você estiver interessado em múltiplos contrastes você pode modificar o parâmetro nlfc, por padrão ele é definido como "1" para que apenas um anel seja desenhado. Os valores logFC são codificados por cores usando uma escala de cores definida pelo usuário (lfc.col); Para ficar bem, o número de canais foi reduzido e a cor dos canais pode ser alterada usando o parâmetro term.col.ainda disponível?GOCluster para ver como alterar os parâmetros. O parâmetro mais importante desta função é cluster.by, que pode ser especificado para agrupar por padrões de expressão gênica ('logFC', como mostrado acima) ou categorias funcionais ('termos').
- GOCluster(circ, EC$process, clust.by = 'term', lfc.col = c('darkgoldenrod1', 'black', 'cyan1'))
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
Os diagramas de Venn podem ser usados para detectar relações entre várias listas de genes expressos diferencialmente ou para explorar a interseção de genes de múltiplas vias em análises funcionais. Os diagramas de Venn não mostram apenas o número de genes sobrepostos, mas também informações sobre o padrão de expressão do gene (geralmente regulado positivamente, frequentemente regulado negativamente ou contra-regulado). Atualmente, até três conjuntos de dados são usados como entrada. Os dados de entrada contêm pelo menos duas colunas: uma para nomes de genes e outra para valores logFC.
- l1 <- subset(circ, term == 'heart development', c(genes,logFC))
- l2 <- subset(circ, term == 'plasma membrane', c(genes,logFC))
- l3 <- subset(circ, term == 'tissue morphogenesis', c(genes,logFC))
- GOVenn(l1,l2,l3, label = c('heart development', 'plasma membrane', 'tissue morphogenesis'))
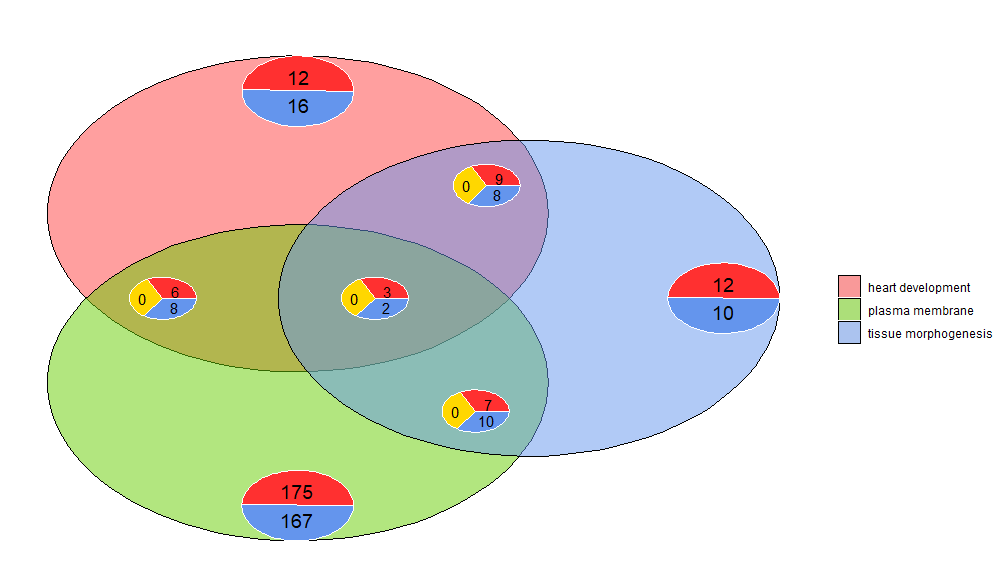
Por exemplo, o desenvolvimento cardíaco e a morfogênese tecidual têm 22 genes, 12 são regulados positivamente e 10 são regulados negativamente. Uma coisa importante a observar é que os gráficos de pizza não exibem informações redundantes. Portanto, se três conjuntos de dados forem comparados, os genes comuns a todos os conjuntos de dados (o gráfico de pizza do meio) não serão incluídos nos outros gráficos de pizza. Esta ferramenta está disponível no brilhanteapp https://wwalter.shinyapps.io/Venn/, a ferramenta web é mais interativa, o círculo possui uma área proporcional ao número de genes no conjunto de dados e o controle deslizante pode ser usado para mover o pequeno gráfico de pizza, e possui GOVenn Features todas as opções para alterar o layout do gráfico e também para baixar imagens e listas de genes.
Página inicial do software: https://wencke.github.io/

Ele se dedica à pesquisa de tecnologia há mais de 30 anos e é proficiente em diversas linguagens como java, linux, javascript, php, css, etc. estação de documentação do desenvolvedor para compartilhar alguns problemas no desenvolvimento de tecnologia para referência futura.

Correspondência[email protected]