
私の連絡先情報
郵便メール:
2024-07-12
한어Русский языкEnglishFrançaisIndonesianSanskrit日本語DeutschPortuguêsΕλληνικάespañolItalianoSuomalainenLatina
GOPlot パッケージは生物学的データの視覚化に使用されます。むしろ、このパッケージは発現データと機能解析の結果を統合し、視覚化します。でも気をつけてねこのパッケージは、結果を視覚化するためだけに使用でき、これらの分析を実行するために使用することはできません。 。科学のあらゆる分野において、スペースの制約と結果に必要な簡潔さのせいで、物事を現実的に説明することは困難であるため、情報を視覚化して画像を使用して情報を伝える必要があります。適切に設計されたグラフィックスにより、少ないスペースでより多くの情報が提供されます。このパッケージのアイデアは、ユーザーが大量のデータを迅速に調査し、データの傾向を明らかにし、データのパターンと相関関係を見つけられるようにすることです。
データの視覚化は、生物学的な疑問に対する答えを見つけたり、特定の仮説を判断したり、さまざまな問題を調査するためのさまざまな角度を発見したりするのに役立ちます。そして、このパッケージのプロット機能は、データ全体から始まり、選択された遺伝子と対応する経路のサブセットで終わる、データの階層構造に基づいて開発されています。
例を挙げて具体的に説明しましょう。
GEO から提供されるデータを GOplot と呼びます。GSE47067、2 つの組織 (脳と心臓) の内皮細胞のトランスクリプトーム情報が含まれています。詳細については、Nolan らの論文 https://www.ncbi.nlm.nih.gov/pubmed/23871589 を参照してください。データは正規化され、差次的に発現される遺伝子が見つかります。、その後、DAVID 関数アノテーション ツールを使用します (DAVID アノテーション データの更新が遅いため、現時点では推奨されていません。使用することをお勧めします)Go East、最高のオンライン GO エンリッチメント分析ツールそしてたった 1 ステップでエンリッチメント分析を実行できるこの Web サイトは、公開される前に CNS などで 350 回以上引用されました。濃縮分析を実行し、GSEA を 1 つの記事でマスターする、超詳細なチュートリアル) 差次的に発現された遺伝子の遺伝子アノテーション (adjusted p-value < 0.05 )および機能強化分析。このデータ セットには、次の 5 つのカテゴリのデータが含まれています。
| 名前 | 説明する | データセットのサイズ |
|---|---|---|
| EC$eset | 脳および心臓内皮細胞における正常化された遺伝子発現 (3 反復) | 20644 x 7 |
| EC$ジェネリスト | 差次的に発現された遺伝子 (調整された p 値 < 0.05) | 2039 x 7 |
| EC$デビッド | DAVIDを用いた差次的遺伝子の機能強化解析の結果 | 174×5 |
| EC$遺伝子 | 遺伝子とlogFC | 37×2 |
| EC$プロセス | 強化された生物学的プロセスのための選択された特徴ベクトル | 7 |
差次的に発現された遺伝子の GO が豊富な経路を確認したいのですが、描画を開始する前に、形式要件を満たすデータを提供する必要があります。基本的にグラフを描くために必要なデータは自分で用意するのですが、このパッケージには機能がありますcircle_datデータ形式の処理に役立ちます。circle_dat主に差次的に発現する遺伝子について、選択した遺伝子の機能強化解析結果とその logFC 値を組み合わせることができます。circle_dat使い方はとても簡単で、2つのデータを読み込むだけです。最初のデータには、機能強化分析の結果が含まれており、少なくとも 4 つの列 (機能強化分析カテゴリ、経路、遺伝子、調整済み p 値) が含まれています。2 番目のデータは選択した遺伝子とその logFC のもので、このデータはソースとして使用できます。limma統計分析の結果 (経歴からの注: 2 つのファイルに必ず注目してください)遺伝子の名前の付け方すべてのように一貫性を保つGene symbol )。例を挙げて上記のデータ形式を見てみましょう。
- #安装已发布的稳定版本
- #install.packages('GOplot')
- #安装github上的开发版本
- #install_github('wencke/wencke.github.io')
- #载入包
- library(GOplot)
- #读入包内自带的数据
- data(EC)
- #查看功能富集分析结果的数据格式
- head(EC$david)
-
- ## Category ID Term
- ## 1 BP GO:0007507 heart development
- ## 2 BP GO:0001944 vasculature development
- ## 3 BP GO:0001568 blood vessel development
- ## 4 BP GO:0048729 tissue morphogenesis
- ## 5 BP GO:0048514 blood vessel morphogenesis
- ## 6 BP GO:0051336 regulation of hydrolase activity
- ## Genes
- ## 1 DLC1, NRP2, NRP1, EDN1, PDLIM3, GJA1, TTN, GJA5, ZIC3, TGFB2, CERKL, GATA6, COL4A3BP, GAB1, SEMA3C, MKL2, SLC22A5, MB, PTPRJ, RXRA, VANGL2, MYH6, TNNT2, HHEX, MURC, MIB1, FOXC2, FOXC1, ADAM19, MYL2, TCAP, EGLN1, SOX9, ITGB1, CHD7, HEXIM1, PKD2, NFATC4, PCSK5, ACTC1, TGFBR2, NF1, HSPG2, SMAD3, TBX1, TNNI3, CSRP3, FOXP1, KCNJ8, PLN, TSC2, ATP6V0A1, TGFBR3, HDAC9
- ## 2 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, EFNB2, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 3 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 4 DLC1, ENAH, NRP1, PGF, ZIC2, TGFB2, CD44, ILK, SEMA3C, RET, AR, RXRA, VANGL2, LEF1, TNNT2, HHEX, MIB1, NCOA3, FOXC2, FOXC1, TGFB1I1, WNT5A, COBL, BBS4, FGFR3, TNC, BMPR2, CTNND1, EGLN1, NR3C1, SOX9, TCF7L1, IGF1R, FOXQ1, MACF1, HOXA5, BCL2, PLXND1, CAR2, ACTC1, TBX4, SMAD3, FZD3, SHANK3, FZD6, HOXB4, FREM2, TSC2, ZIC5, TGFBR3, APAF1
- ## 5 GNA13, CAV1, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, CDH2, MEIS1, WT1, TGFB2, WARS, PTK2, CERKL, APOE, CXCR4, ANG, SEMA3C, PLCD1, NOS2, MKL2, PLXND1, FIGF, FGF2, PTPRJ, TGFBR2, TBX4, NF1, TBX1, TNNI3, PNPLA6, VASH1, THY1, NUS1, MEOX2, ECSCR, AMOT, HBEGF, FOXC2, FOXC1, HDAC7
- ## 6 CAV1, XIAP, AGFG1, ADORA2A, TNNC1, TBC1D9, LEPR, ABHD5, EDN1, ASAP2, ASAP3, SMAP1, TBC1D12, ANG, TBC1D14, MTCH1, TBC1D13, TBC1D4, TBC1D30, DHCR24, HIP1, VAV3, NOS1, NF1, MYH6, RICTOR, TBC1D22A, THY1, PLCE1, RNF7, NDEL1, CHML, IFT57, ACAP2, TSC2, ERN1, APAF1, ARAP3, ARAP2, ARAP1, HTR2A, F2R
- ## adj_pval
- ## 1 0.000002170
- ## 2 0.000010400
- ## 3 0.000007620
- ## 4 0.000119000
- ## 5 0.000720000
- ## 6 0.001171166
-
- #查看基因的数据格式
- head(EC$genelist)
-
- ## ID logFC AveExpr t P.Value adj.P.Val B
- ## 1 Slco1a4 6.645388 1.2168670 88.65515 1.32e-18 2.73e-14 29.02715
- ## 2 Slc19a3 6.281525 1.1600468 69.95094 2.41e-17 2.49e-13 27.62917
- ## 3 Ddc 4.483338 0.8365231 65.57836 5.31e-17 3.65e-13 27.18476
- ## 4 Slco1c1 6.469384 1.3558865 59.87613 1.62e-16 8.34e-13 26.51242
- ## 5 Sema3c 5.515630 2.3252117 58.53141 2.14e-16 8.81e-13 26.33626
- ## 6 Slc38a3 4.761755 0.9218670 54.11559 5.58e-16 1.76e-12 25.70308
2 つの入力データ形式を理解すると、次のように使用できます。cirlce_dat描画データを生成する機能です。
- # 生成画图所需的数据格式
- circ <- circle_dat(EC$david, EC$genelist)
-
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
circオブジェクトには 8 列のデータがあります。
カテゴリ: BP (生物学的プロセス)、CC (細胞成分) または MF (分子機能)
ID: GO id (オプションの列。GO id に基づいていない機能分析ツールを使用する場合は、ID 列を選択できません。ここでの ID は KEGG ID にすることもできます)
用語: GO 経路
count: 各経路の遺伝子の数
gene: 遺伝子名 - logFC: 各遺伝子のlogFC値
adj_pval: 調整された p 値、adj_pval<0.05 の経路は有意に濃縮されていると見なされます。
zscore: zscore は統計的正規化方法を指すものではありませんが、生物学的プロセス (/分子機能/細胞成分) が減少する可能性が高いか (負の値)、増加する可能性が高いか (正の値) を推定するために簡単に計算される値です。計算方法は、上方制御される遺伝子の数から下方制御される遺伝子の数を引いたものを、各経路の遺伝子数の平方根で割ったものです。
最初にデータを確認するときは、グラフからできるだけ多くのパスウェイを表示し、価値のあるパスウェイを見つけたいと考えています。そのため、重要性を評価するためのパラメーターが必要です。棒グラフはサンプル データを説明するためによく使用されるため、GOBar 関数を使用すると見栄えの良い棒グラフをすばやく作成できます。
まず、単純な棒グラフを直接生成します。横軸は次のとおりです。GO Terms、彼らによるとzscoreバーを並べ替えると、縦軸は次のようになります。-log(adj p-value);色が表すのはzscore、青色はz-score負の値の場合、対応する経路の遺伝子発現が減少する可能性が高く、赤色で示されます。z-scoreが正の値である場合、対応する経路の遺伝子発現が増加する可能性が高くなります。必要に応じて、パラメータ order.by.zscore を FALSE に設定することで順序を変更できます。この場合、バーは重要度に基づいて順序付けされます。
- # 生成简单的条形图
- GOBar(subset(circ, category == 'BP'))
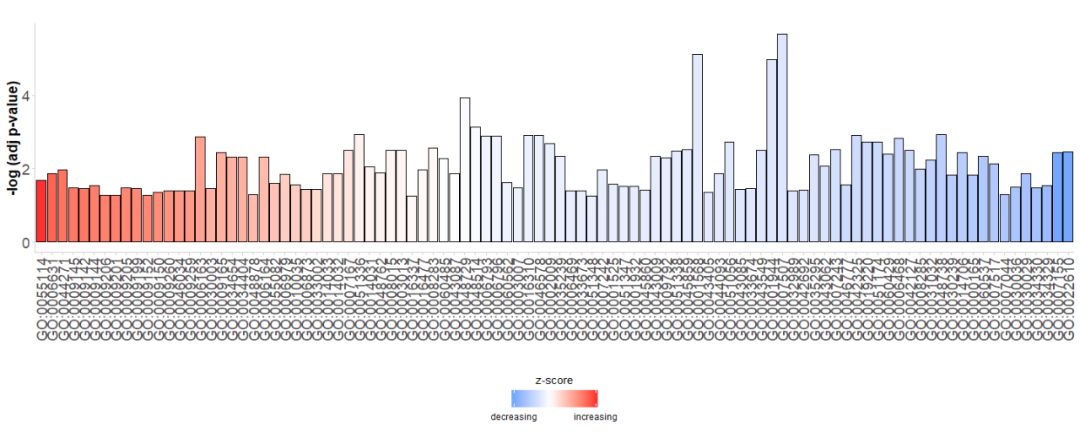
#GOBar(subset(circ, category == 'BP',order.by.zscore=FALSE))さらに、チャネルのカテゴリに応じて棒グラフを描画するように表示パラメータを変更します。
- #根据通路的类别来绘制条形图
- GOBar(circ, display = 'multiple')
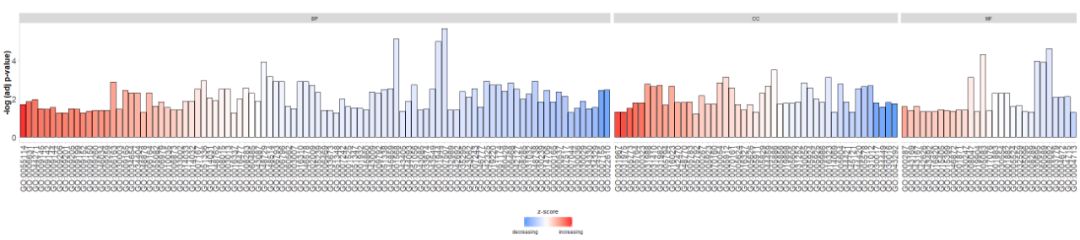
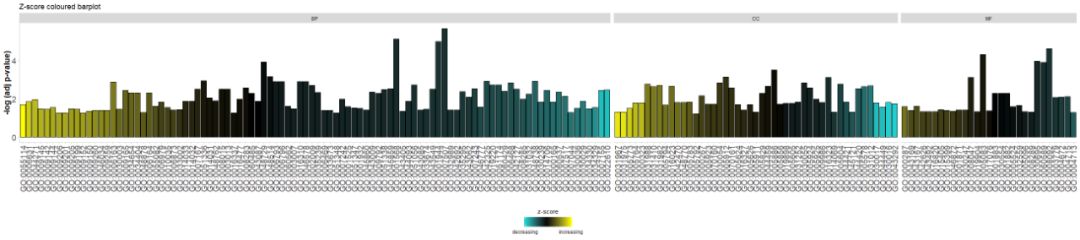
タイトルを追加してパラメータを使用するzsc.col変化zscoreの色。
- # Facet the barplot, add a title and change the colour scale for the z-score
- GOBar(circ, display = 'multiple', title = 'Z-score coloured barplot', zsc.col = c('yellow', 'black', 'cyan'))
棒グラフは非常に一般的で理解しやすいですが、バブル チャートを使用すると、データに関するより多くの情報を表示できます。
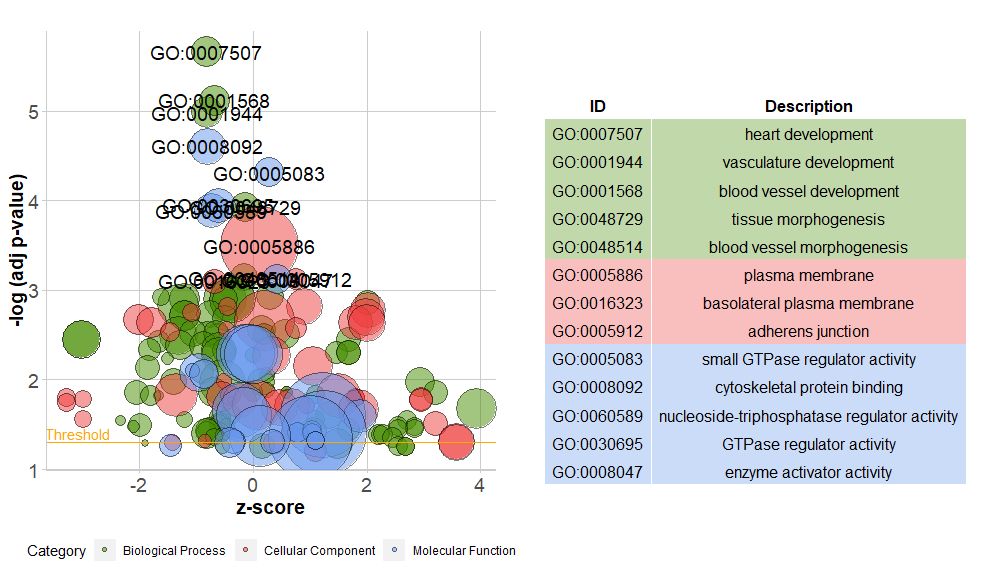
横軸はzscore;縦軸は-log(adj p-value)棒グラフと同様に、値が高いほど、円の面積は対応する経路内の遺伝子の数に関連しています。circ$count ); 色は経路に対応するカテゴリに対応し、緑色は生物学的プロセス、赤色は細胞成分、青色は分子機能を示します。入力できるのは、?GOBubble画像のパラメータをすべて変更するには、GOBubble 関数のヘルプ ページを参照してください。デフォルトでは、各円には対応する GO ID がマークされており、右側には GO ID と GO 用語の対応関係を示す表も表示されます。パラメータは次のように設定できます。table.legendのためにFALSEそれを隠すために。パスの説明を表示したい場合は、パラメータ ID を FALSE に設定します。ただし、スペースが限られており、円が重なっているため、すべての円がマークされるわけではなく、円のみがマークされます。-log(adj p-value) > 3(デフォルトは5)。
- # 生成泡泡图,并展示-log(adj p-value) > 3 的通路的GO ID
- GOBubble(circ, labels = 3)
バブル チャートにタイトルを追加するか、円の色を指定して各カテゴリのパスウェイを個別に表示し、表示される GO ID しきい値を変更する場合は、次のパラメータを追加できます。
GOBubble(circ, title = 'Bubble plot', colour = c('orange', 'darkred', 'gold'), display = 'multiple', labels = 3)
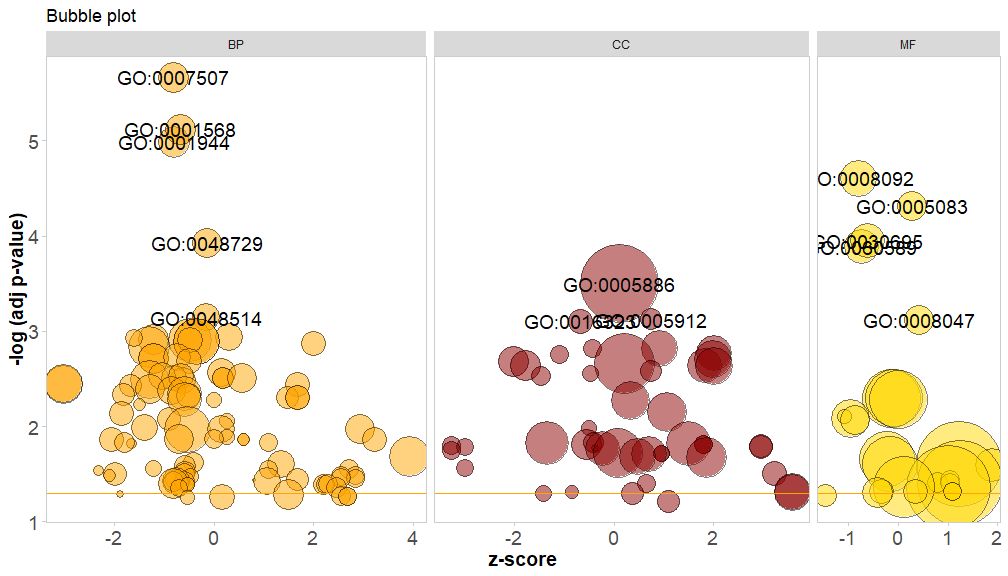
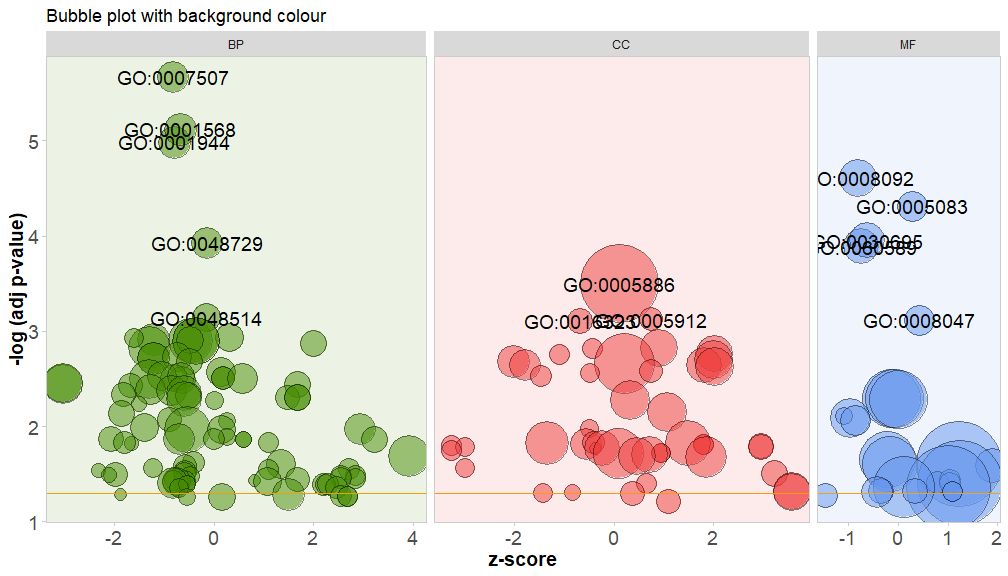
パラメータ bg.col を TRUE に設定して、チャネルのクラスの背景に色を付けます。
GOBubble(circ, title = 'Bubble plot with background colour', display = 'multiple', bg.col = T, labels = 3)
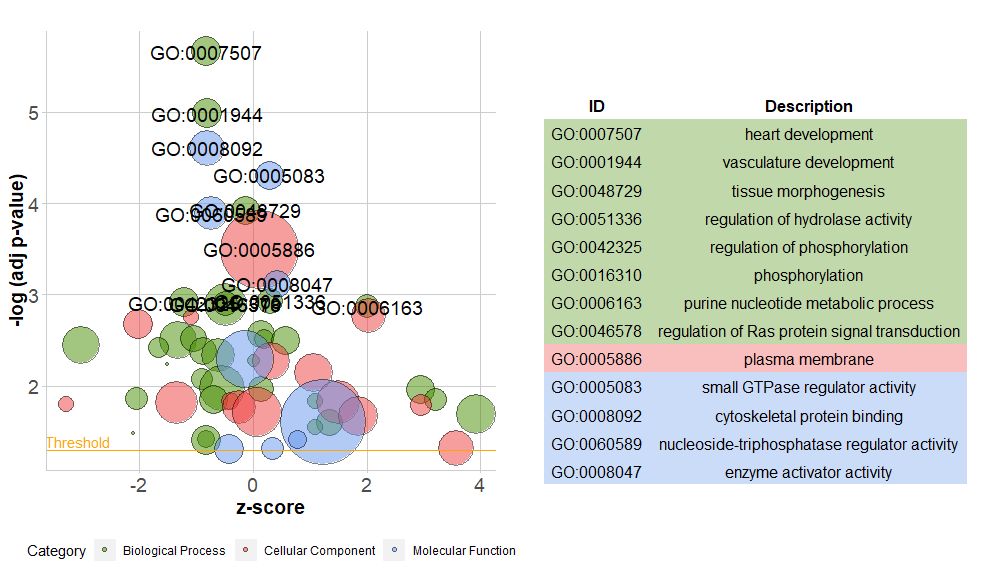
新しいバージョンのパッケージには新しい機能が含まれていますreduce_overlap 、この機能は、冗長な項目の数を減らすことができます。つまり、遺伝子の重複が設定されたしきい値以上であるすべての経路を削除し、すべての項目の表示に関係なく、各グループから 1 つの経路のみを代表として保持できます。 GO の経路。冗長な項の数を減らすことにより、プロット (バブル プロットなど) の読みやすさが大幅に向上します。
- # reduce_overlap,参数设置为0.75
- reduced_circ <- reduce_overlap(circ, overlap = 0.75)
-
- GOBubble(reduced_circ, labels = 2.8)
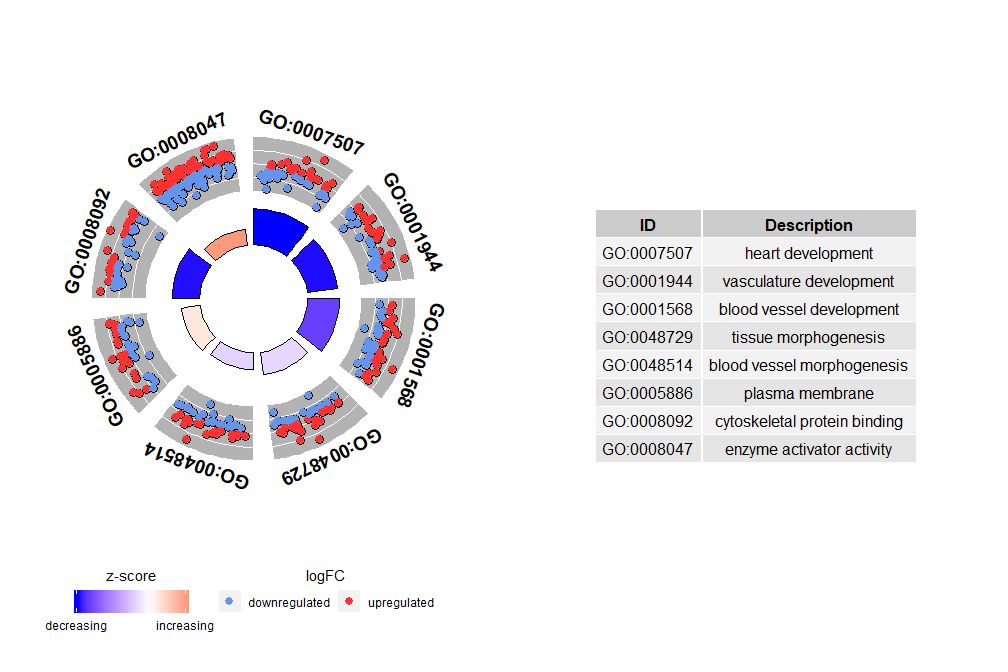
すべての情報を示すグラフは、どの経路が最も意味があるかを発見するのに役立ちますが、実際はデータで確認したい仮説やアイデアに依存しており、最も重要な経路が必ずしも関心のある経路であるとは限りません。したがって、貴重な経路のセットを手動で選択する場合 (EC$process )、この特定の一連の経路に関するより詳細な情報を示す図が必要です。しかし、これらの数字を提示すると問題が発生します。解釈が難しい場合があります。zscore情報提供。結局のところ、この計算方法は普遍的ではありません。上方制御された遺伝子の数から下方制御された遺伝子の数を引いた値を、各経路の遺伝子数の平方根で割ったものです。GOCircle結果のグラフもこの事実を強調しています。
円図の外側の円は、各経路の遺伝子のlogFC値を点として示している。赤い丸はアップレギュレーションを示し、青はダウンレギュレーションを示します。パラメータが使用可能lfc.col色を変える。これは、場合によっては非常に重要な経路の z スコアがゼロに近い理由も説明します。 zscore が 0 であっても、チャネルが重要ではないという意味ではありません。これは、zscore が大まかな尺度であることを示しているだけです。なぜなら、zscore は生物学的プロセスにおける個々の遺伝子の機能レベルと活性化依存性も考慮していないからです。
GOCircle(circ)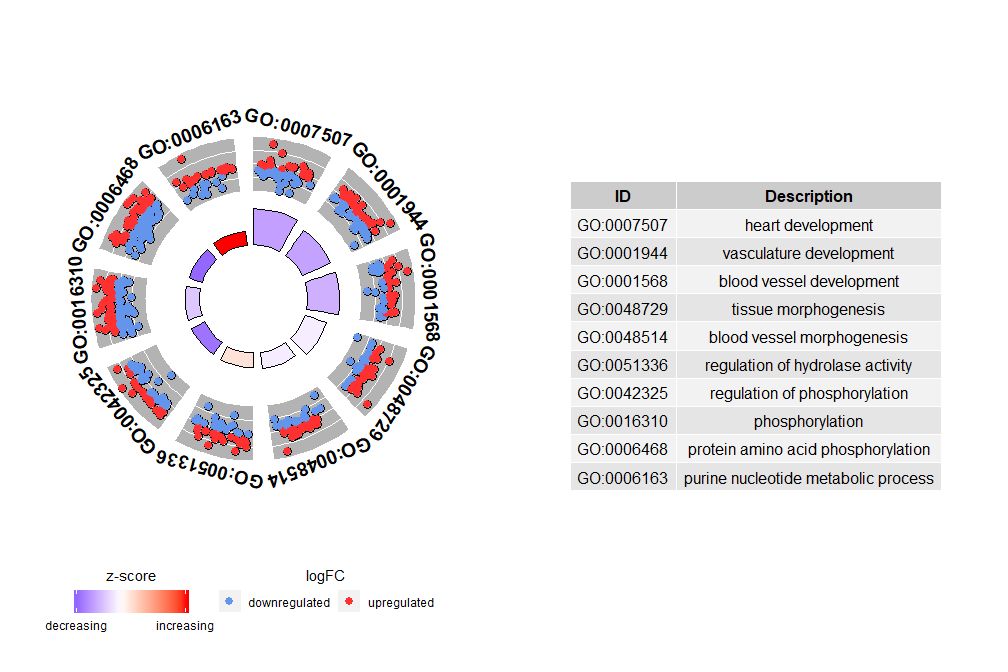
nsubパラメーターには数値または文字ベクトルを設定できます。文字ベクトルの場合は、表示する GO ID またはパスウェイが含まれます。
- # 生成特定通路的圈图
- IDs <- c('GO:0007507', 'GO:0001568', 'GO:0001944', 'GO:0048729', 'GO:0048514', 'GO:0005886', 'GO:0008092', 'GO:0008047')
- GOCircle(circ, nsub = IDs)
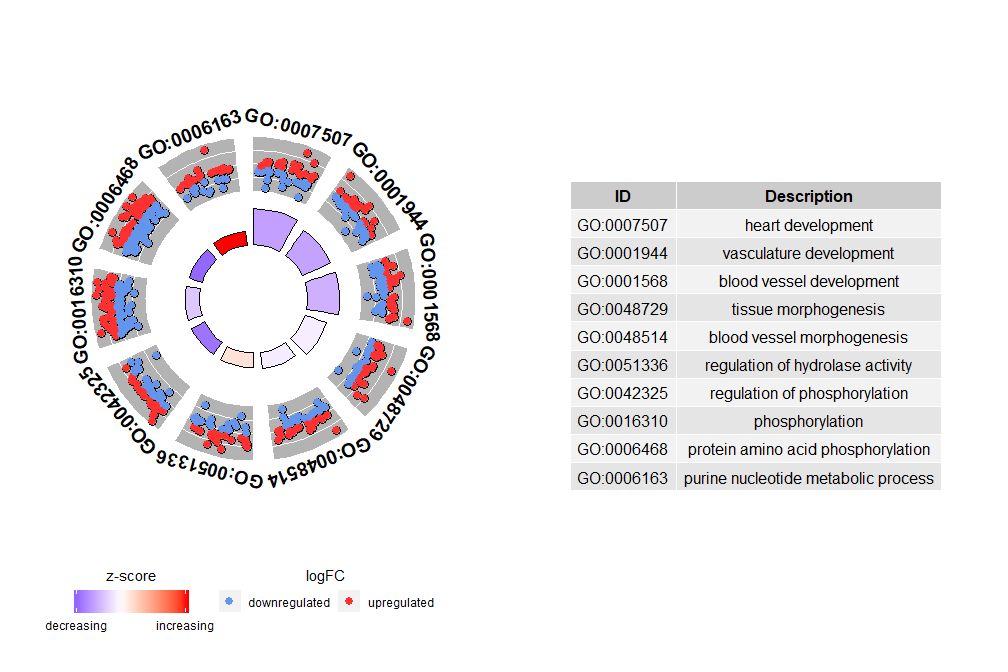
nsub が数値ベクトルの場合、その数値は表示する数値を定義します。入力データフレームの最初の行から始まります。この視覚化は、より小さいデータでのみ機能します。最大チャネル数のデフォルトは 12 です。チャンネル数は減りますが、表示される情報量は増えます。
- # 圈图展示数据前十个通路
- GOCircle(circ, nsub = 10)
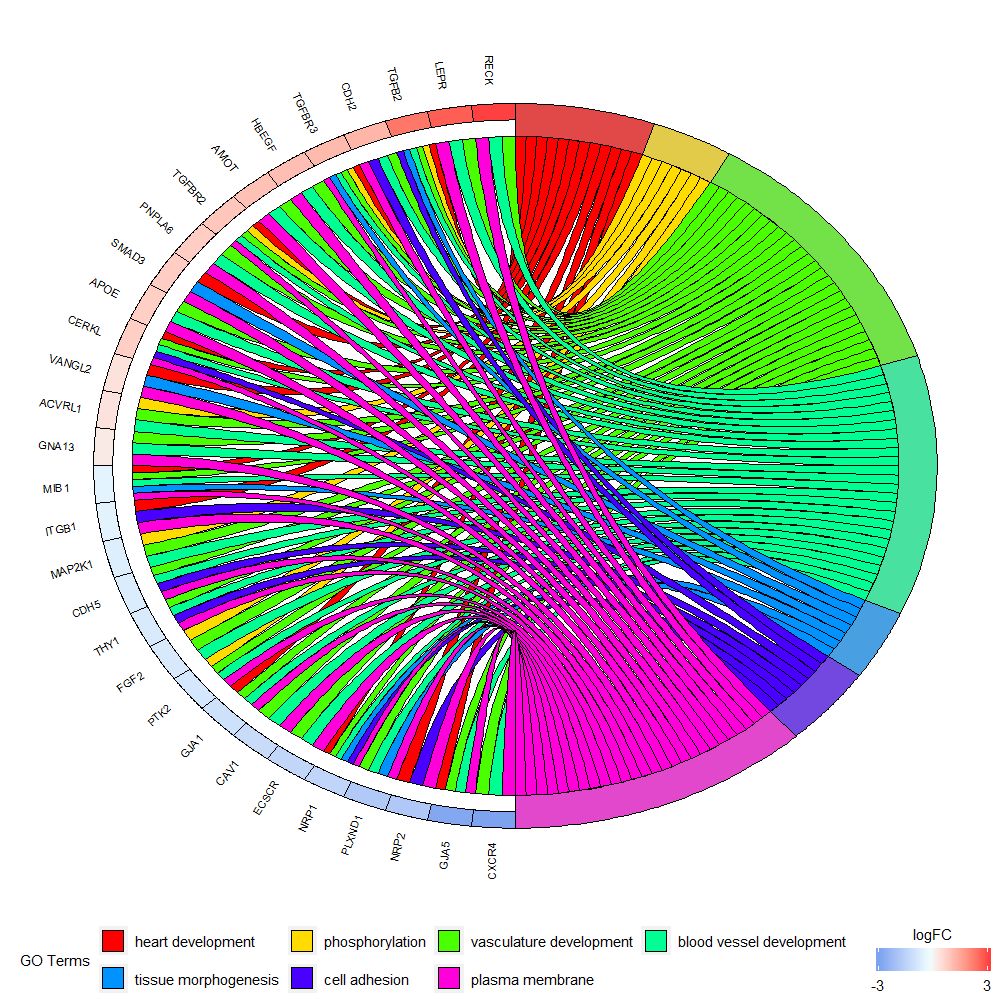
GOChord は、選択した遺伝子と経路の関係と遺伝子の logFC を表示できます。まず、自分で構築できる行列を入力する必要があります。0-1マトリックス、関数も使用できますchord_dat構築します。この関数にはデータ、遺伝子、プロセスの 3 つのパラメータがあり、最後の 2 つのパラメータには少なくとも 1 つのパラメータが必要です。それから関数circle_dat発現データと機能解析の結果を組み合わせます。
棒グラフとバブル チャートは、データの第一印象を与えることができます。GOCircle は、パスウェイ内の遺伝子の発現値を表示するレイヤーを追加しますが、情報が 1 つもありません。遺伝子と複数の経路の間の関係について。特定の遺伝子が複数のプロセスに関連しているかどうかを解明するのは簡単ではありません。 GOChord は GOCircle の欠点を補います。生成されたデータの行は遺伝子、列は経路です。「0」は遺伝子が経路に割り当てられていないことを意味し、「1」はその逆です。
- # 找到感兴趣的的基因,这里我们以EC$genes为例
- head(EC$genes)
-
- ## ID logFC
- ## 1 PTK2 -0.6527904
- ## 2 GNA13 0.3711599
- ## 3 LEPR 2.6539788
- ## 4 APOE 0.8698346
- ## 5 CXCR4 -2.5647537
- ## 6 RECK 3.6926860
-
- # 获得感兴趣基因的通路
- EC$process
-
- ## [1] "heart development" "phosphorylation"
- ## [3] "vasculature development" "blood vessel development"
- ## [5] "tissue morphogenesis" "cell adhesion"
- ## [7] "plasma membrane"
-
- # 使用chord_dat构建矩阵
- chord <- chord_dat(circ, EC$genes, EC$process)
- head(chord)
-
- ## heart development phosphorylation vasculature development
- ## PTK2 0 1 1
- ## GNA13 0 0 1
- ## LEPR 0 0 1
- ## APOE 0 0 1
- ## CXCR4 0 0 1
- ## RECK 0 0 1
- ## blood vessel development tissue morphogenesis cell adhesion
- ## PTK2 1 0 0
- ## GNA13 1 0 0
- ## LEPR 1 0 0
- ## APOE 1 0 0
- ## CXCR4 1 0 0
- ## RECK 1 0 0
- ## plasma membrane logFC
- ## PTK2 1 -0.6527904
- ## GNA13 1 0.3711599
- ## LEPR 1 2.6539788
- ## APOE 1 0.8698346
- ## CXCR4 1 -2.5647537
- ## RECK 1 3.6926860
この例では、genes パラメーターのみを指定した場合、結果は、選択された遺伝子と、少なくとも 1 つの指定された遺伝子を含むすべてのプロセス構築のリストになります。0-1マトリックス; 指定された場合のみprocessパラメータを指定すると、結果としてすべての遺伝子が生成されます。0-1リスト内の少なくとも 1 つのプロセスに割り当てられた遺伝子のマトリックス。遺伝子とプロセス パラメーターのみを指定すると、非常に大きな 0 ~ 1 の行列が生成され、視覚化結果が混乱する可能性があることに注意してください。
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
-
- # Generate the matrix with a list of selected genes
- chord_genes <- chord_dat(data = circ, genes = EC$genes)
- head(chord_genes)
-
- ## heart development vasculature development blood vessel development
- ## PTK2 0 1 1
- ## GNA13 0 1 1
- ## LEPR 0 1 1
- ## APOE 0 1 1
- ## CXCR4 0 1 1
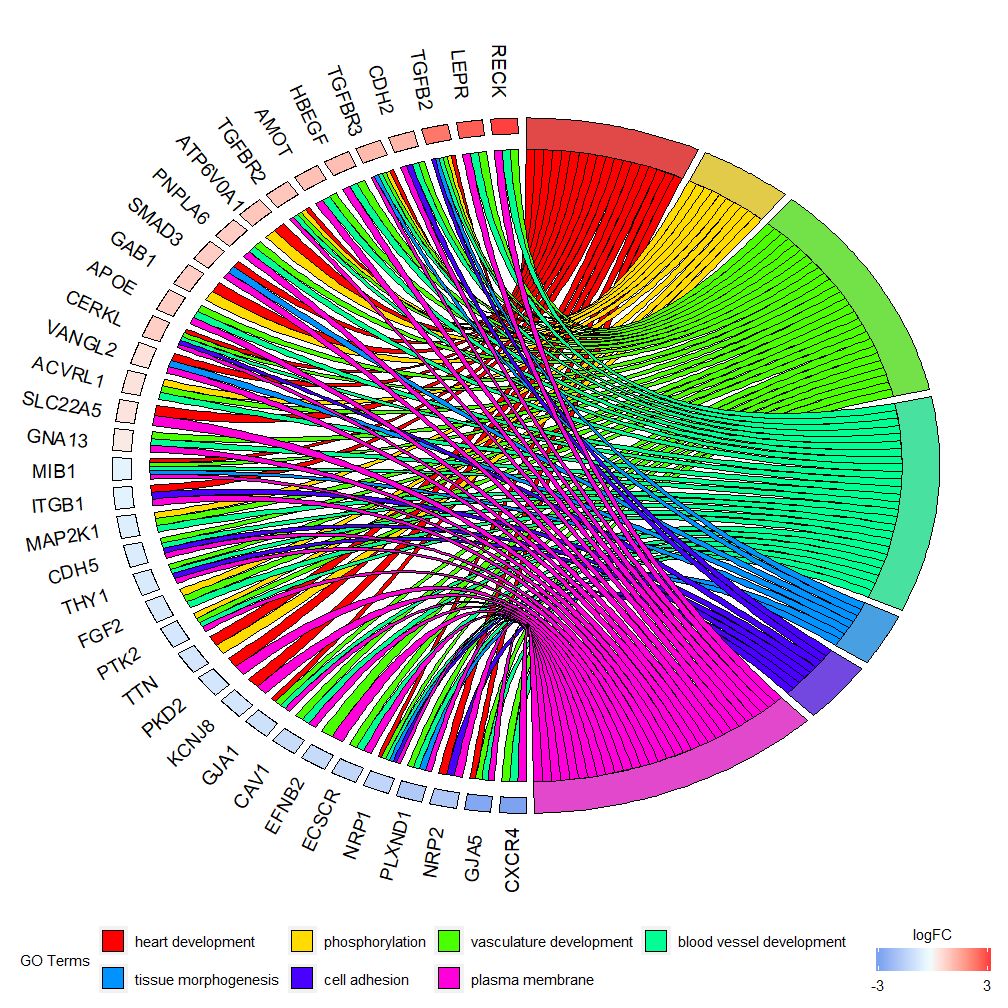
このチャートは、高次元データのより小さなサブセットを示すことを目的としています。調整できるパラメータは主に 2 つあります。gene.orderそしてnlfc 。 遺伝子パラメータは、「logFC」、「alphabetical」、「none」として指定できます。実際、一般に遺伝子パラメータを logFC として指定します。nlfc パラメータは、各遺伝子が行列内に 0 個以上の logFC 値をどのように持つかを処理できるため、この関数の最も重要なパラメータの 1 つです。したがって、エラーを避けるためにパラメータを指定する必要があります。
たとえば、logFC 値のない行列がある場合は、次のように設定する必要があります。nlfc=0 ; または、複数の条件またはバッチで遺伝子の差次的発現解析を実行する場合、各遺伝子には複数の logFC 値が含まれるため、nlfc=logFC 列番号を設定する必要があります。ほとんどの場合、遺伝子ごとに logFC 値が 1 つだけ存在すると考えられるため、デフォルト値は「1」です。 space パラメーターを使用して、logFC を表す色付きの四角形間のスペースを定義します。 gene.size パラメータは遺伝子名のフォント サイズを指定し、gene.space パラメータは遺伝子名の間のスペース サイズを指定します。
- chord <- chord_dat(data = circ, genes = EC$genes, process = EC$process)
- GOChord(chord, space = 0.02, gene.order = 'logFC', gene.space = 0.25, gene.size = 5)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
logFC値に応じて設定可能gene.order=‘logFC’ 、遺伝子は logFC 値に従って並べ替えられます。場合によっては、画像が少し混雑することがありますが、これは、limit パラメーターを使用して表示される遺伝子または経路の数を減らすことで自動化できます。 Limit は 2 つのカットオフ値を持つベクトルです (デフォルトは c(0,0))。最初の値は、遺伝子を割り当てる必要がある経路の最小数を指定します。 2 番目の値は、経路に割り当てられる遺伝子の数を決定します。
- # 仅显示分配给至少三个通路的基因
- GOChord(chord, limit = c(3, 0), gene.order = 'logFC')
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
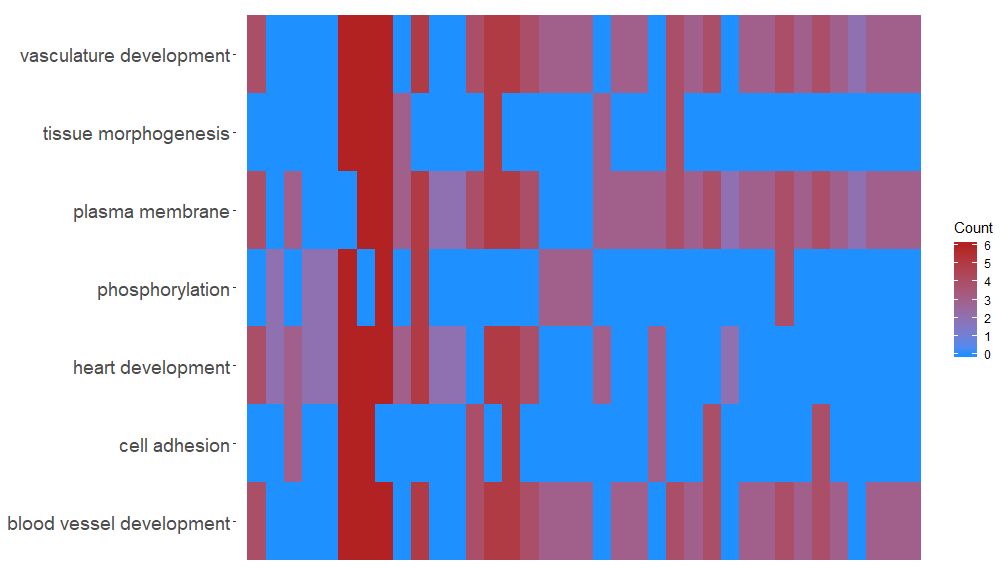
GOHeat機能はGOChordと同様に遺伝子とパスウェイの関係をヒートマップで表示することができます。生物学的プロセスは水平方向に表示され、遺伝子は垂直方向に表示されます。各列は小さな長方形に分割されており、色は通常、logFC 値に応じて異なります。さらに、同様の機能経路が豊富に含まれる遺伝子がクラスター化されました。ヒートマップのカラー選択には、nlfc パラメーターに応じて 2 つのモードがあります。 nlfc = 0 の場合、色は各遺伝子の濃縮された経路の数です。詳細については、例を参照してください。
- # First, we use the chord object without logFC column to create the heatmap
- GOHeat(chord[,-8], nlfc = 0)
GOHeat(chord[,-8])色はnlfc = 1の場合の遺伝子のlogFCに対応します。
GOHeat(chord, nlfc = 1, fill.col = c('red', 'yellow', 'green'))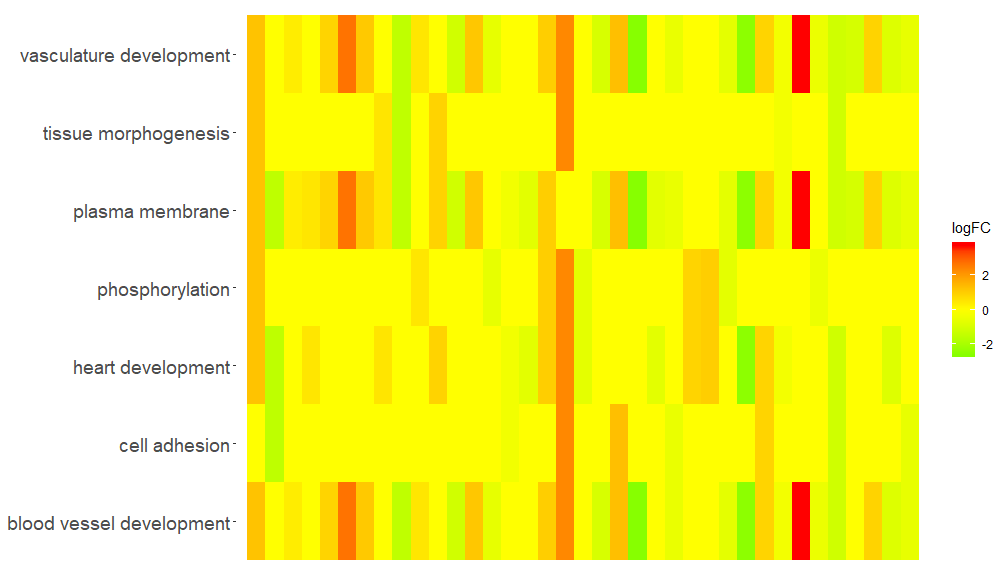
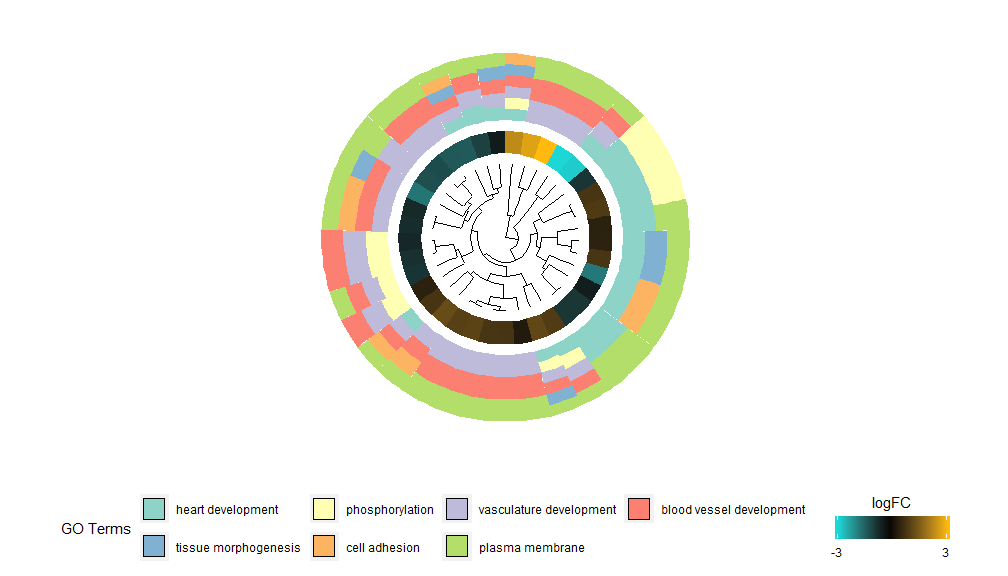
GOCluster 機能の背後にある考え方は、できるだけ多くの情報を表示することです。以下に例を示します。
- GOCluster(circ, EC$process, clust.by = 'logFC', term.width = 2)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
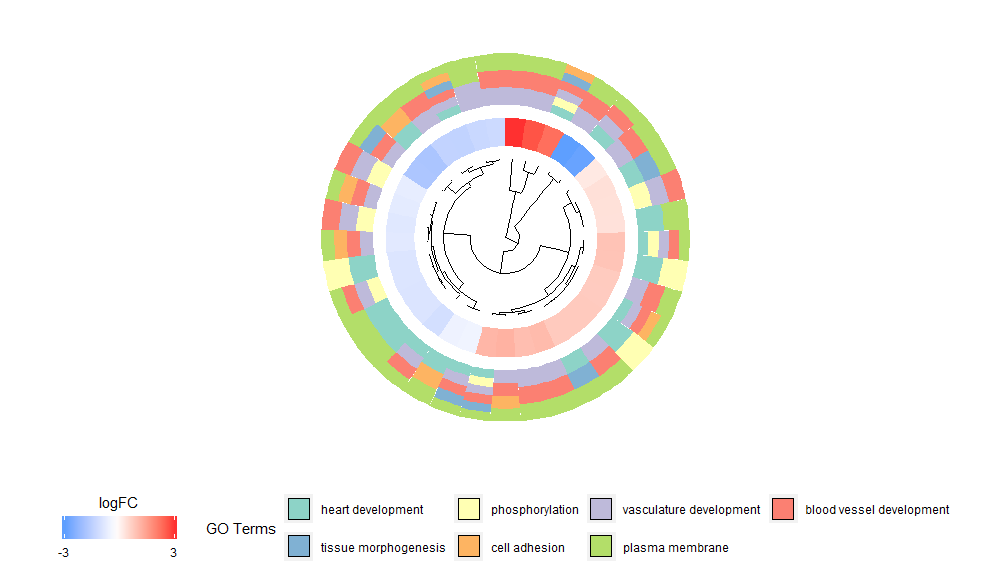
階層的クラスタリングは、遺伝子発現に関する一般的な教師なしクラスタリング分析方法であり、発現パターンによって遺伝子を公平にグループ化することを保証するため、クラスターには、共制御される遺伝子または機能的に関連する遺伝子の複数のグループが含まれる可能性があります。 GOCluster はhclustこの方法は、遺伝子発現プロファイルの階層的クラスタリングを実行します。距離メトリックまたはクラスタリング アルゴリズムを変更する場合は、それぞれ metric および clust パラメーターを使用します。結果の樹状図は、ggdendro を使用して変換し、ggplot2 で視覚化できます。効果的であるだけでなく、視覚的にも魅力的な円形レイアウトを選択してください。樹状図の隣の最初の円は遺伝子の logFC を表しており、これは実際にはクラスタリング ツリーのリーフです。複数のコントラストに興味がある場合は、nlfc パラメータを変更できます。デフォルトでは、リングが 1 つだけ描画されるように「1」に設定されています。 logFC 値は、ユーザー定義可能なカラー スケール (lfc.col) を使用して色分けされています。次の円は、遺伝子に割り当てられた経路を表します。見栄えを良くするために、チャネルの数が減らされており、パラメータ term.col を使用してチャネルの色を変更できます。まだ利用可能です?GOClusterパラメータを変更する方法を確認します。この関数の最も重要なパラメータは、cluster.by です。これは、遺伝子発現パターン (上記の「logFC」) または機能カテゴリ (「用語」) によってクラスター化するように指定できます。
- GOCluster(circ, EC$process, clust.by = 'term', lfc.col = c('darkgoldenrod1', 'black', 'cyan1'))
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
ベン図を使用すると、差次的に発現する遺伝子のさまざまなリスト間の関係を検出したり、機能解析で複数の経路遺伝子の交差を探索したりできます。ベン図は、重複する遺伝子の数を示すだけでなく、遺伝子の発現パターン (通常は上方制御され、多くの場合は下方制御または逆制御) に関する情報も示します。現在、最大 3 つのデータセットが入力として使用されます。入力データには少なくとも 2 つの列が含まれます。1 つは遺伝子名用、もう 1 つは logFC 値用です。
- l1 <- subset(circ, term == 'heart development', c(genes,logFC))
- l2 <- subset(circ, term == 'plasma membrane', c(genes,logFC))
- l3 <- subset(circ, term == 'tissue morphogenesis', c(genes,logFC))
- GOVenn(l1,l2,l3, label = c('heart development', 'plasma membrane', 'tissue morphogenesis'))
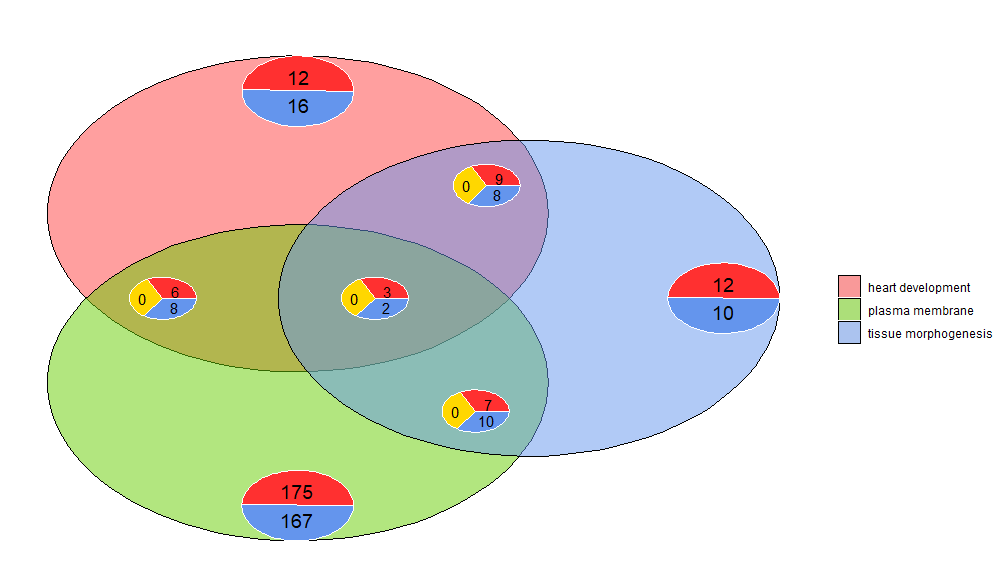
たとえば、心臓の発生と組織の形態形成には 22 個の遺伝子があり、12 個は上方制御され、10 個は下方制御されます。注意すべき重要な点は、円グラフには冗長な情報が表示されないことです。したがって、3 つのデータセットを比較すると、すべてのデータセット (中央の円グラフ) に共通する遺伝子は他の円グラフには含まれません。このツールは Shinyapp https://wwalter.shinyapps.io/Venn/ で利用できます。Web ツールはよりインタラクティブで、円の面積はデータセット内の遺伝子の数に比例し、スライダーを使用してオブジェクトを移動できます。小さな円グラフで、プロットのレイアウトを変更したり、画像や遺伝子リストをダウンロードしたりするための GOVenn 機能のすべてのオプションがあります。
ソフトウェアホームページ: https://wencke.github.io/

彼は 30 年以上テクノロジーの研究に専念しており、java、linux、javascript、php、css などのさまざまな言語に堪能であり、オープンソース分野で多くの貢献を行っています。将来の参考のために技術開発におけるいくつかの問題を共有する開発者ドキュメント ステーション。

郵便メール: