2024-07-12
한어Русский языкEnglishFrançaisIndonesianSanskrit日本語DeutschPortuguêsΕλληνικάespañolItalianoSuomalainenLatina
Le package GOPlot est utilisé pour la visualisation de données biologiques. Au contraire, ce package intègre et visualise les données d'expression avec les résultats des analyses fonctionnelles.Mais fais attentionCe package ne peut pas être utilisé pour effectuer ces analyses, uniquement pour visualiser les résultats. . Dans tous les domaines scientifiques, il est difficile de décrire les choses de manière réaliste en raison des contraintes d'espace et de la simplicité requise pour les résultats. Les informations doivent donc être visualisées et des images utilisées pour transmettre l'information. Des graphiques bien conçus fournissent plus d'informations dans moins d'espace. L'idée du package est de permettre aux utilisateurs d'examiner rapidement de grandes quantités de données, de révéler des tendances dans les données et de trouver des modèles et des corrélations dans les données.
La visualisation des données peut nous aider à trouver des réponses à des questions biologiques, à juger une certaine hypothèse et même à découvrir différents angles pour étudier différents problèmes. Et les fonctions de traçage de ce package sont développées sur la base de la structure hiérarchique des données, en commençant par les données globales et en terminant par un sous-ensemble de gènes sélectionnés et les voies correspondantes.
Expliquons-le concrètement avec un exemple.
Nous appelons les données fournies avec GOplot, qui proviennent de GEOGSE47067, contenant des informations sur le transcriptome des cellules endothéliales de deux tissus (cerveau et cœur). Pour plus d'informations, consultez l'article de Nolan et al. https://www.ncbi.nlm.nih.gov/pubmed/23871589, puis.Les données sont normalisées et des gènes différentiellement exprimés sont trouvés., puis utilisez l'outil d'annotation de fonction DAVID (les données d'annotation DAVID sont mises à jour lentement et ne sont pas recommandées pour le moment. Il est recommandé d'utiliserGo East, le meilleur outil d'analyse d'enrichissement GO en ligneetCe site Web, qui permet d'effectuer une analyse d'enrichissement en une seule étape, a été cité plus de 350 fois par CNS et d'autres avant sa publication.Effectuer une analyse d'enrichissement,Maîtrisez GSEA en un seul article, tutoriel super détaillé) Annotation génétique de gènes exprimés différentiellement (adjusted p-value < 0.05 ) et analyse d'enrichissement fonctionnel. Cet ensemble de données contient les cinq catégories de données suivantes :
| nom | décrire | Taille de l'ensemble de données |
|---|---|---|
| EC$eset | Expression génique normalisée dans les cellules endothéliales du cerveau et du cœur (3 répétitions) | 20644 x 7 |
| EC$généliste | Gènes exprimés différentiellement (valeur p ajustée <0,05) | 2039 x 7 |
| EC$david | Résultats de l'analyse d'enrichissement fonctionnel de gènes différentiels à l'aide de DAVID | 174 x 5 |
| Gène EC$ | Gènes et logFC | 37 x 2 |
| Processus EC$ | Vecteurs de caractéristiques sélectionnés pour des processus biologiques enrichis | 7 |
Nous voulons voir les voies enrichies par GO des gènes exprimés différentiellement, mais avant de commencer à dessiner, nous devons fournir des données qui répondent aux exigences de format.De manière générale, les données nécessaires au tracé du graphique sont fournies par vous-même, maisIl y a une fonction dans ce packagecircle_datPeut nous aider à gérer le format des données。circle_datIl peut combiner les résultats de l’analyse d’enrichissement fonctionnel de gènes sélectionnés et leurs valeurs logFC, principalement pour les gènes différentiellement exprimés.circle_dat L'utilisation est très simple, il suffit de lire deux données. Les premières données contiennent les résultats de l’analyse d’enrichissement fonctionnel, avec au moins quatre colonnes (catégorie d’analyse d’enrichissement fonctionnel, voie, gène, valeur p ajustée).La deuxième donnée concerne le gène sélectionné et son logFC, ces données peuvent être la sourcelimmaLes résultats de l'analyse statistique (Note des biographies : assurez-vous de prêter attention à deux fichiersComment les gènes sont nommésSoyez cohérent, comme tousGene symbol ). Examinons les formats de données mentionnés ci-dessus avec des exemples.
- #安装已发布的稳定版本
- #install.packages('GOplot')
- #安装github上的开发版本
- #install_github('wencke/wencke.github.io')
- #载入包
- library(GOplot)
- #读入包内自带的数据
- data(EC)
- #查看功能富集分析结果的数据格式
- head(EC$david)
-
- ## Category ID Term
- ## 1 BP GO:0007507 heart development
- ## 2 BP GO:0001944 vasculature development
- ## 3 BP GO:0001568 blood vessel development
- ## 4 BP GO:0048729 tissue morphogenesis
- ## 5 BP GO:0048514 blood vessel morphogenesis
- ## 6 BP GO:0051336 regulation of hydrolase activity
- ## Genes
- ## 1 DLC1, NRP2, NRP1, EDN1, PDLIM3, GJA1, TTN, GJA5, ZIC3, TGFB2, CERKL, GATA6, COL4A3BP, GAB1, SEMA3C, MKL2, SLC22A5, MB, PTPRJ, RXRA, VANGL2, MYH6, TNNT2, HHEX, MURC, MIB1, FOXC2, FOXC1, ADAM19, MYL2, TCAP, EGLN1, SOX9, ITGB1, CHD7, HEXIM1, PKD2, NFATC4, PCSK5, ACTC1, TGFBR2, NF1, HSPG2, SMAD3, TBX1, TNNI3, CSRP3, FOXP1, KCNJ8, PLN, TSC2, ATP6V0A1, TGFBR3, HDAC9
- ## 2 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, EFNB2, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 3 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 4 DLC1, ENAH, NRP1, PGF, ZIC2, TGFB2, CD44, ILK, SEMA3C, RET, AR, RXRA, VANGL2, LEF1, TNNT2, HHEX, MIB1, NCOA3, FOXC2, FOXC1, TGFB1I1, WNT5A, COBL, BBS4, FGFR3, TNC, BMPR2, CTNND1, EGLN1, NR3C1, SOX9, TCF7L1, IGF1R, FOXQ1, MACF1, HOXA5, BCL2, PLXND1, CAR2, ACTC1, TBX4, SMAD3, FZD3, SHANK3, FZD6, HOXB4, FREM2, TSC2, ZIC5, TGFBR3, APAF1
- ## 5 GNA13, CAV1, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, CDH2, MEIS1, WT1, TGFB2, WARS, PTK2, CERKL, APOE, CXCR4, ANG, SEMA3C, PLCD1, NOS2, MKL2, PLXND1, FIGF, FGF2, PTPRJ, TGFBR2, TBX4, NF1, TBX1, TNNI3, PNPLA6, VASH1, THY1, NUS1, MEOX2, ECSCR, AMOT, HBEGF, FOXC2, FOXC1, HDAC7
- ## 6 CAV1, XIAP, AGFG1, ADORA2A, TNNC1, TBC1D9, LEPR, ABHD5, EDN1, ASAP2, ASAP3, SMAP1, TBC1D12, ANG, TBC1D14, MTCH1, TBC1D13, TBC1D4, TBC1D30, DHCR24, HIP1, VAV3, NOS1, NF1, MYH6, RICTOR, TBC1D22A, THY1, PLCE1, RNF7, NDEL1, CHML, IFT57, ACAP2, TSC2, ERN1, APAF1, ARAP3, ARAP2, ARAP1, HTR2A, F2R
- ## adj_pval
- ## 1 0.000002170
- ## 2 0.000010400
- ## 3 0.000007620
- ## 4 0.000119000
- ## 5 0.000720000
- ## 6 0.001171166
-
- #查看基因的数据格式
- head(EC$genelist)
-
- ## ID logFC AveExpr t P.Value adj.P.Val B
- ## 1 Slco1a4 6.645388 1.2168670 88.65515 1.32e-18 2.73e-14 29.02715
- ## 2 Slc19a3 6.281525 1.1600468 69.95094 2.41e-17 2.49e-13 27.62917
- ## 3 Ddc 4.483338 0.8365231 65.57836 5.31e-17 3.65e-13 27.18476
- ## 4 Slco1c1 6.469384 1.3558865 59.87613 1.62e-16 8.34e-13 26.51242
- ## 5 Sema3c 5.515630 2.3252117 58.53141 2.14e-16 8.81e-13 26.33626
- ## 6 Slc38a3 4.761755 0.9218670 54.11559 5.58e-16 1.76e-12 25.70308
Après avoir compris les deux formats de données d'entrée, vous pouvez utilisercirlce_datfonction pour générer des données de dessin.
- # 生成画图所需的数据格式
- circ <- circle_dat(EC$david, EC$genelist)
-
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
circL'objet comporte huit colonnes de données, à savoir
catégorie : BP (Processus Biologique), CC (Composant Cellulaire) ou MF (Fonction Moléculaire)
ID : GO id (colonne facultative, si vous souhaitez utiliser un outil d'analyse fonctionnelle qui n'est pas basé sur GO id, vous ne pouvez pas sélectionner la colonne ID ; l'ID ici peut également être KEGG ID)
terme : voie GO
count : nombre de gènes dans chaque voie
gène : nom du gène - logFC : valeur logFC de chaque gène
adj_pval : valeur p ajustée, les voies avec adj_pval <0,05 sont considérées comme significativement enrichies
zscore : zscore ne fait pas référence à une méthode de normalisation statistique, mais est une valeur facilement calculée pour estimer si un processus biologique (/fonction moléculaire/composant cellulaire) est plus susceptible de diminuer (valeur négative) ou d'augmenter (valeur positive).La méthode de calcul est le nombre de gènes régulés positivement moins le nombre de gènes régulés négativement divisé par la racine carrée du nombre de gènes dans chaque voie.
Lorsque nous examinons pour la première fois les données, nous souhaitons afficher autant de voies que possible à partir du graphique, et nous voulons également trouver des voies intéressantes, nous avons donc besoin de certains paramètres pour évaluer leur importance. Les graphiques à barres sont souvent utilisés pour décrire des exemples de données. Nous pouvons donc utiliser la fonction GOBar pour créer rapidement un graphique à barres attrayant.
Tout d’abord, un simple graphique à barres est généré directement. L’axe horizontal est.GO Terms, Selon euxzscoreTriez les barres ; l'axe vertical est-log(adj p-value);La couleur représentezscore, le bleu indiquez-scoreest une valeur négative, l'expression du gène dans la voie correspondante est plus susceptible de diminuer, indiquée en rougez-score est une valeur positive, l’expression des gènes dans la voie correspondante est plus susceptible d’augmenter. Si vous le souhaitez, l'ordre peut être modifié en définissant le paramètre order.by.zscore sur FALSE, auquel cas les barres sont classées en fonction de leur importance.
- # 生成简单的条形图
- GOBar(subset(circ, category == 'BP'))
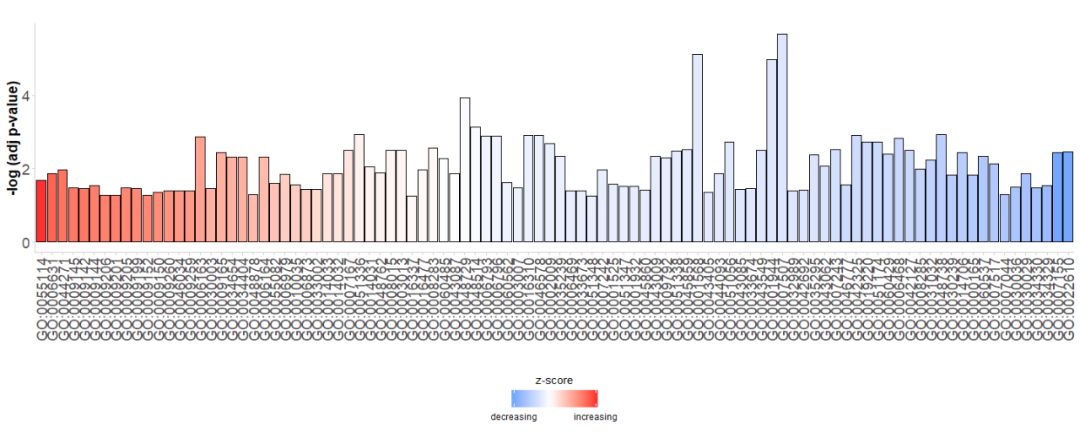
#GOBar(subset(circ, category == 'BP',order.by.zscore=FALSE))De plus, modifiez le paramètre d'affichage pour dessiner un graphique à barres en fonction de la catégorie du canal.
- #根据通路的类别来绘制条形图
- GOBar(circ, display = 'multiple')
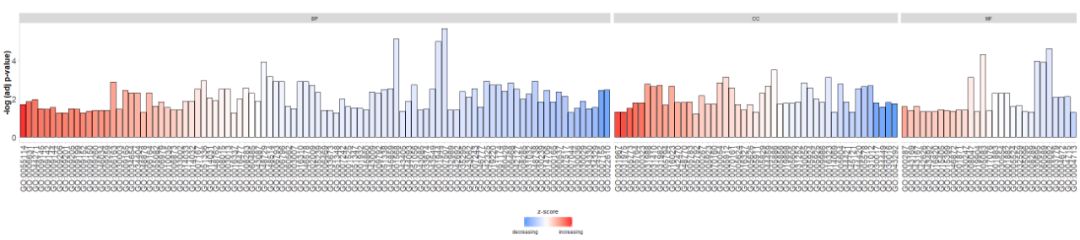
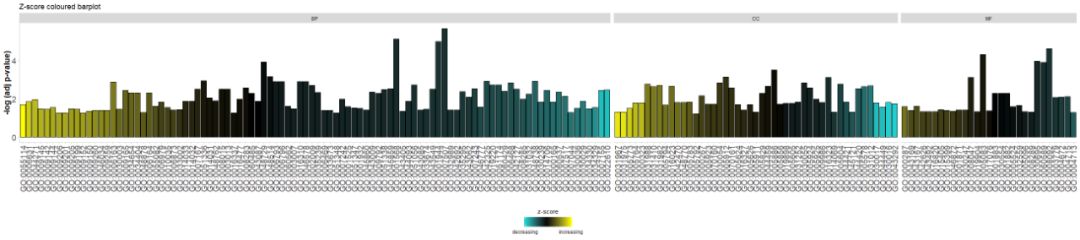
Ajouter un titre et utiliser les paramètreszsc.colChangementzscorela couleur.
- # Facet the barplot, add a title and change the colour scale for the z-score
- GOBar(circ, display = 'multiple', title = 'Z-score coloured barplot', zsc.col = c('yellow', 'black', 'cyan'))
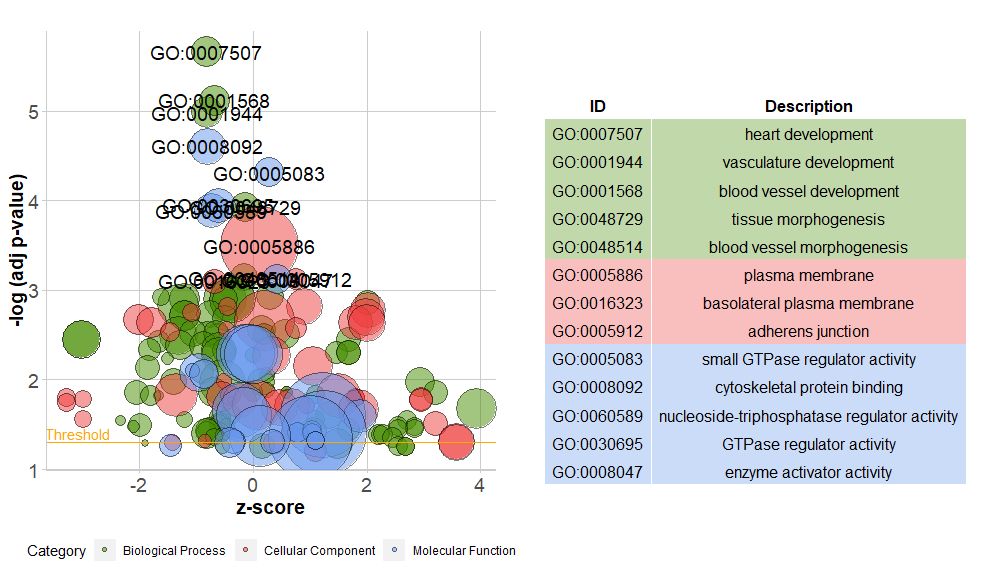
Les graphiques à barres sont très courants et faciles à comprendre, mais nous pouvons utiliser des graphiques à bulles pour afficher plus d'informations sur les données.
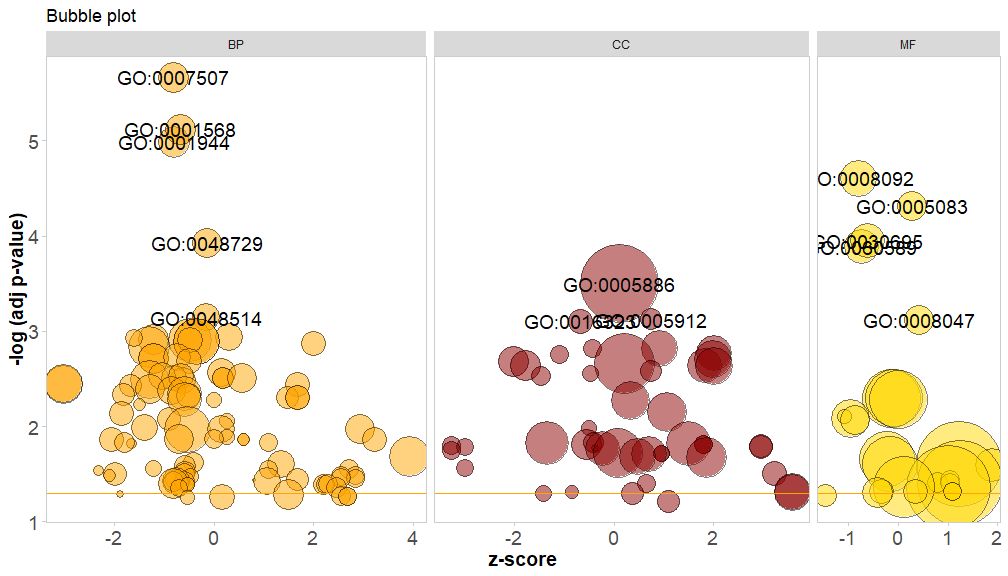
L'axe horizontal estzscore;L'axe vertical est-log(adj p-value), semblable à un histogramme, plus il est élevé, plus l'enrichissement est important ; l'aire du cercle est liée au nombre de gènes dans la voie correspondante (circ$count ); la couleur correspond à la catégorie correspondant à la voie, le vert est le processus biologique, le rouge est la composante cellulaire et le bleu est la fonction moléculaire.Peut être saisi par?GOBubble Consultez la page d'aide de la fonction GOBubble pour modifier tous les paramètres de l'image. Par défaut, chaque cercle est marqué d'un GO ID correspondant, et un tableau montrant la relation correspondante entre GO ID et GO terme est également affiché sur la droite.Les paramètres peuvent être définis partable.legendpourFALSE pour le cacher. Si vous souhaitez afficher la description du chemin, définissez l'ID du paramètre sur FALSE.Cependant, en raison de l'espace limité et du chevauchement des cercles, tous les cercles ne sont pas marqués, seuls les-log(adj p-value) > 3(la valeur par défaut est 5).
- # 生成泡泡图,并展示-log(adj p-value) > 3 的通路的GO ID
- GOBubble(circ, labels = 3)
Si vous souhaitez ajouter un titre au graphique à bulles, ou spécifier la couleur du cercle et afficher les parcours de chaque catégorie séparément et modifier le seuil GO ID affiché, vous pouvez ajouter les paramètres suivants :
GOBubble(circ, title = 'Bubble plot', colour = c('orange', 'darkred', 'gold'), display = 'multiple', labels = 3)
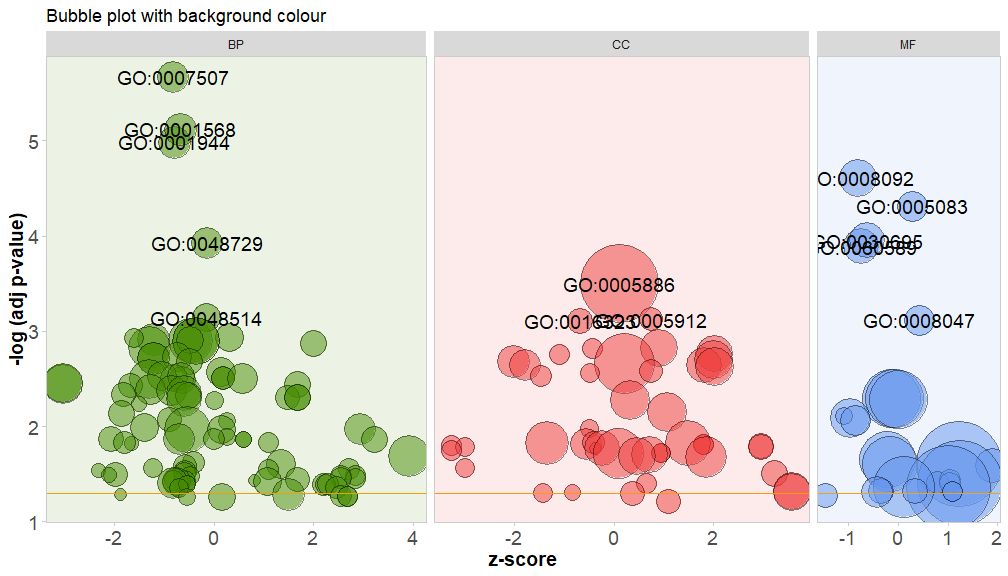
Colorez l'arrière-plan de la classe du canal en définissant le paramètre bg.col sur TRUE.
GOBubble(circ, title = 'Bubble plot with background colour', display = 'multiple', bg.col = T, labels = 3)
La nouvelle version du package contient une nouvelle fonctionreduce_overlap , cette fonction peut réduire le nombre d'éléments redondants, c'est-à-dire qu'elle peut supprimer toutes les voies dont le chevauchement génétique est supérieur ou égal au seuil défini, et ne conserver qu'une seule voie de chaque groupe en tant que représentative, quel que soit l'affichage de toutes. voies dans GO. En réduisant le nombre de termes redondants, la lisibilité des tracés (tels que les tracés à bulles) est considérablement améliorée.
- # reduce_overlap,参数设置为0.75
- reduced_circ <- reduce_overlap(circ, overlap = 0.75)
-
- GOBubble(reduced_circ, labels = 2.8)
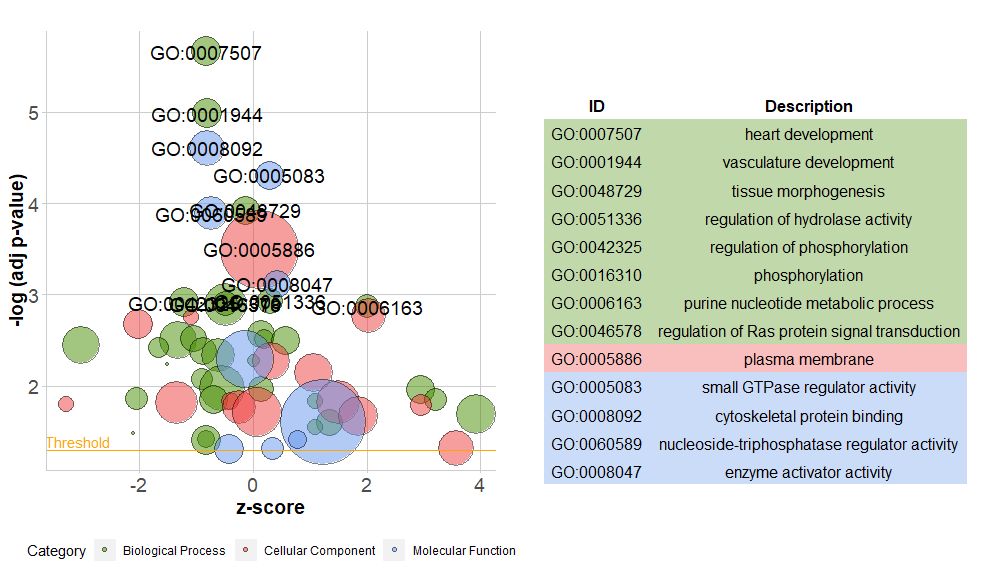
Même si un graphique montrant toutes les informations peut nous aider à découvrir quelles voies sont les plus significatives, la réalité dépend toujours des hypothèses et des idées que vous souhaitez confirmer avec les données, et les voies les plus importantes ne sont pas nécessairement celles qui vous intéressent. Par conséquent, en sélectionnant manuellement un ensemble précieux de voies (EC$process ), nous avons besoin d’un diagramme pour nous montrer des informations plus détaillées sur cet ensemble spécifique de voies.Mais un problème se pose dans la présentation de ces chiffres : il est parfois difficile d'interpréterzscore Informations fournies.Après tout, cette méthode de calcul n’est pas universelle. Comme indiqué ci-dessus, il s’agit simplement du nombre de gènes régulés positivement moins le nombre de gènes régulés négativement divisé par la racine carrée du nombre de gènes dans chaque voie, en utilisantGOCircleLe graphique résultant souligne également ce fait.
Le cercle extérieur du diagramme circulaire montre la valeur logFC des gènes de chaque voie sous forme de points dispersés. Les cercles rouges indiquent une régulation positive et les bleus une régulation négative.Les paramètres peuvent être utiliséslfc.col Changer de couleur. Cela explique également pourquoi, dans certains cas, des parcours très importants ont des zscores proches de zéro. Un zscore de zéro ne signifie pas que le canal est sans importance. Cela montre simplement que le zscore est une mesure approximative, car évidemment, le zscore ne prend pas non plus en compte le niveau fonctionnel et la dépendance à l'activation des gènes individuels dans les processus biologiques.
GOCircle(circ)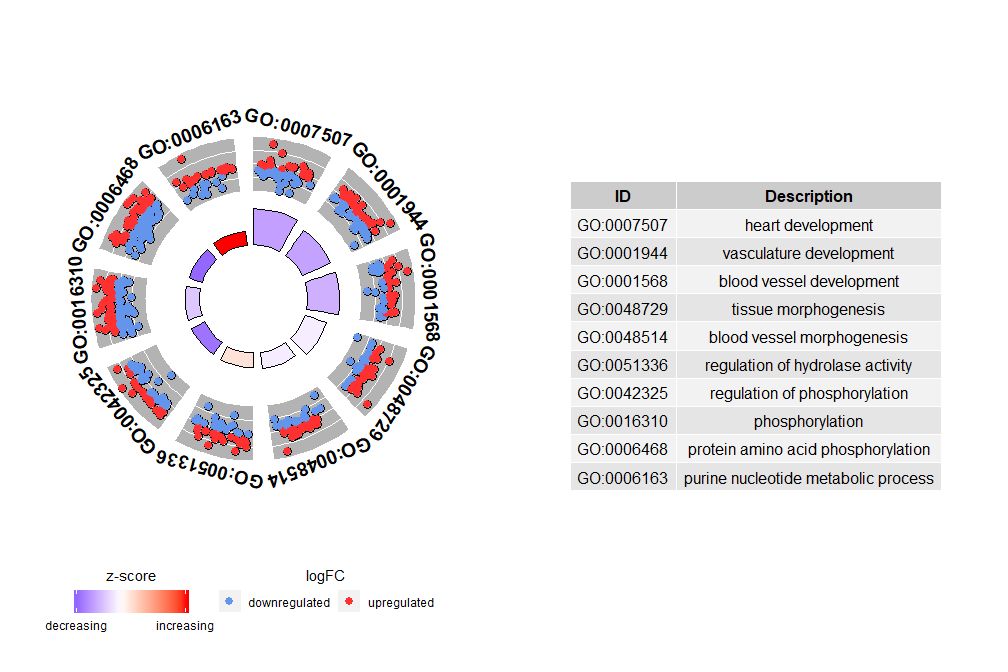
nsub Les paramètres peuvent être des nombres définis ou des vecteurs de caractères. S'il s'agit d'un vecteur de caractères, il contient le GO ID ou le chemin à afficher ;
- # 生成特定通路的圈图
- IDs <- c('GO:0007507', 'GO:0001568', 'GO:0001944', 'GO:0048729', 'GO:0048514', 'GO:0005886', 'GO:0008092', 'GO:0008047')
- GOCircle(circ, nsub = IDs)
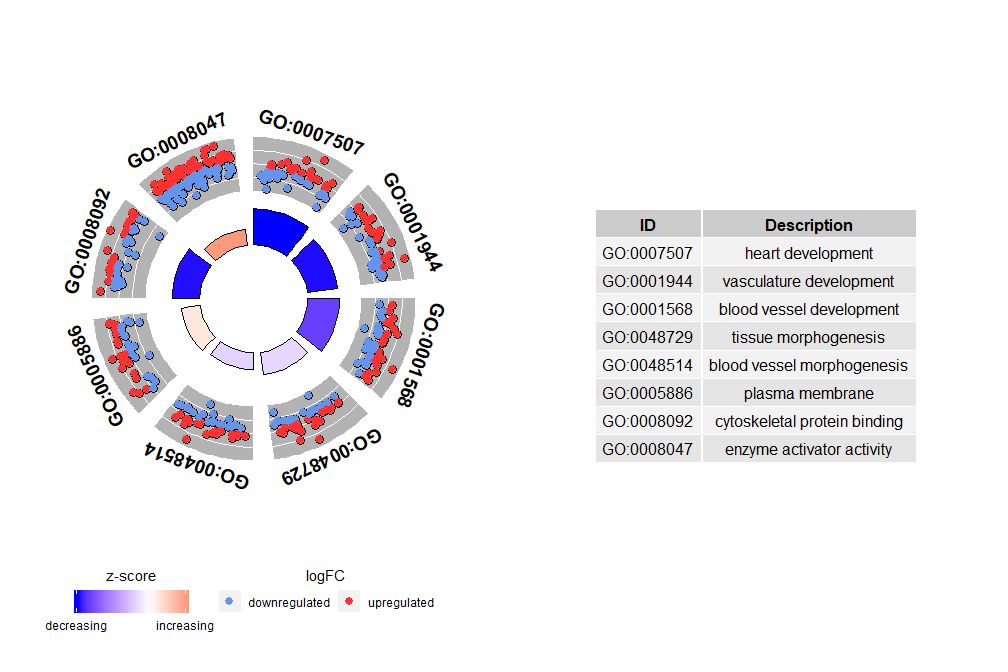
Si nsub est un vecteur numérique, le nombre définit le nombre à afficher. Cela commence à partir de la première ligne du bloc de données d’entrée. Cette visualisation ne fonctionne qu'avec des données plus petites. Le nombre maximum de canaux est par défaut de 12. Bien que le nombre de canaux soit réduit, la quantité d'informations affichées augmente.
- # 圈图展示数据前十个通路
- GOCircle(circ, nsub = 10)
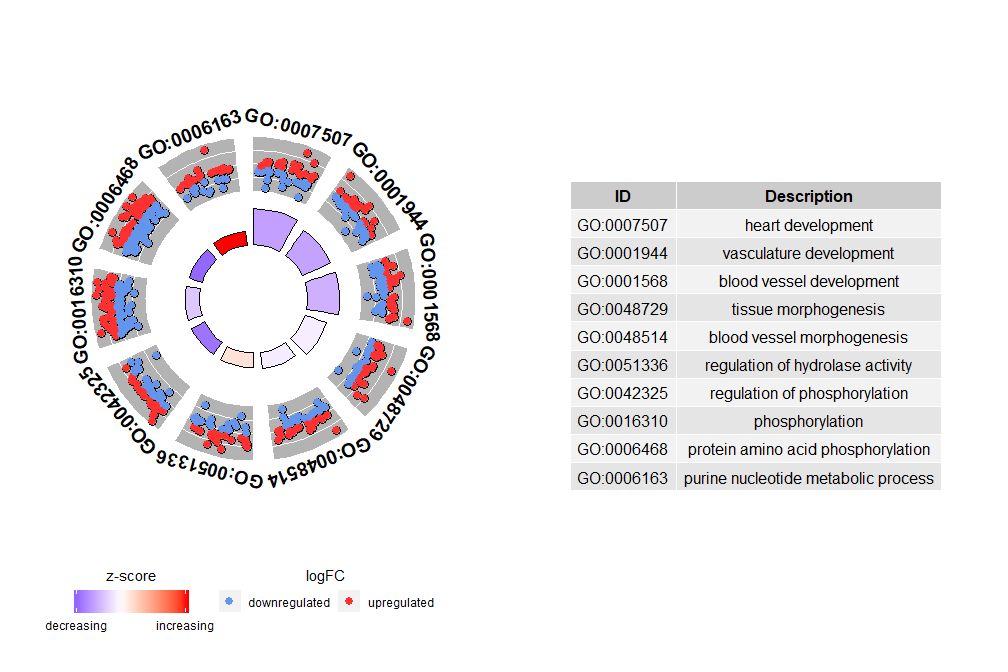
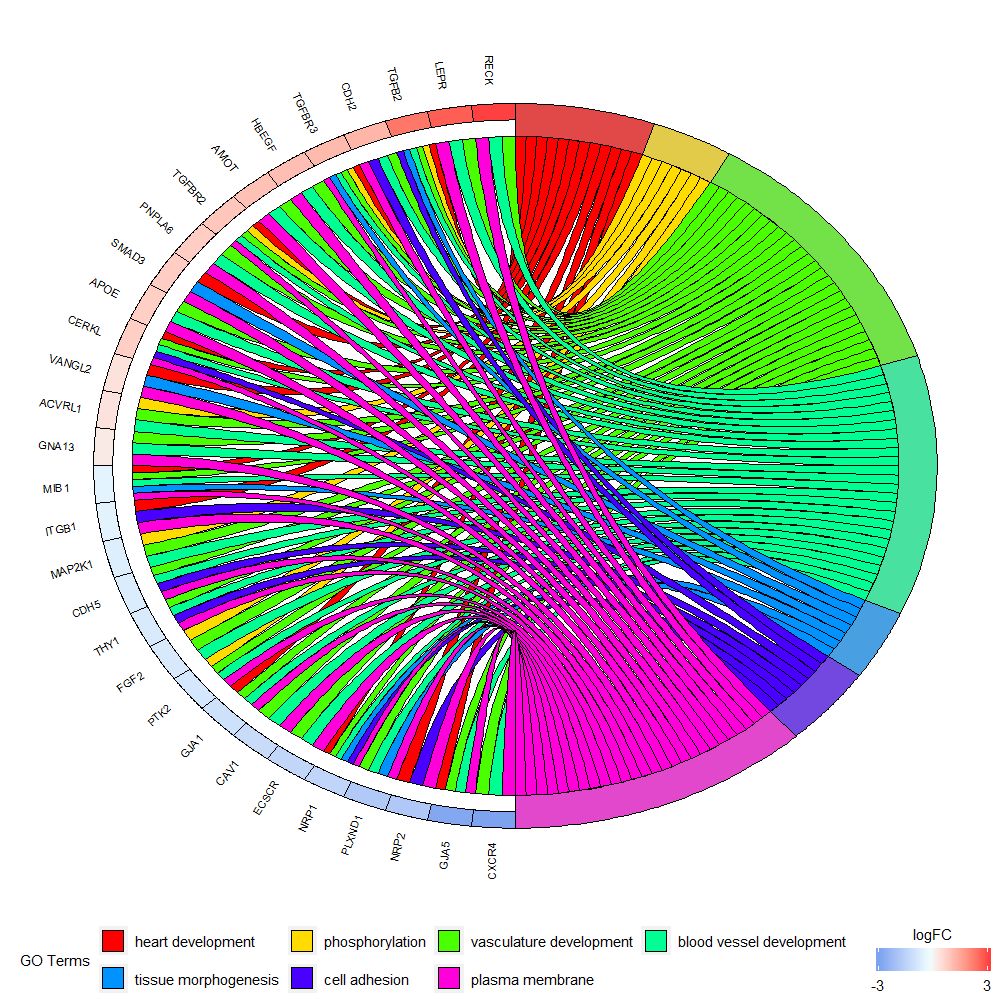
GOChord peut afficher la relation entre les gènes et les voies sélectionnés et le logFC des gènes.Vous devez d’abord saisir une matrice que vous pouvez construire vous-même.0-1Matrix, vous pouvez également utiliser des fonctionschord_dat Construction. Cette fonction comporte trois paramètres : données, gènes et processus, dont les deux derniers paramètres doivent avoir au moins un paramètre.Alors la fonctioncircle_datCombinez les données d’expression avec les résultats des analyses fonctionnelles.
Les graphiques à barres et les graphiques à bulles peuvent vous donner une première impression des données. Désormais, vous pouvez sélectionner certains gènes et voies que nous pensons utiles. Bien que GOCircle ajoute une couche pour afficher la valeur d'expression des gènes dans les voies, il manque des informations individuelles sur. relations entre les gènes et les voies multiples. Il n'est pas facile de déterminer si certains gènes sont liés à plusieurs processus. GOChord compense les défauts de GOCircle. Les lignes des données générées sont des gènes et les colonnes sont des voies. « 0 » signifie que le gène n'est pas affecté à la voie, et « 1 » est le contraire.
- # 找到感兴趣的的基因,这里我们以EC$genes为例
- head(EC$genes)
-
- ## ID logFC
- ## 1 PTK2 -0.6527904
- ## 2 GNA13 0.3711599
- ## 3 LEPR 2.6539788
- ## 4 APOE 0.8698346
- ## 5 CXCR4 -2.5647537
- ## 6 RECK 3.6926860
-
- # 获得感兴趣基因的通路
- EC$process
-
- ## [1] "heart development" "phosphorylation"
- ## [3] "vasculature development" "blood vessel development"
- ## [5] "tissue morphogenesis" "cell adhesion"
- ## [7] "plasma membrane"
-
- # 使用chord_dat构建矩阵
- chord <- chord_dat(circ, EC$genes, EC$process)
- head(chord)
-
- ## heart development phosphorylation vasculature development
- ## PTK2 0 1 1
- ## GNA13 0 0 1
- ## LEPR 0 0 1
- ## APOE 0 0 1
- ## CXCR4 0 0 1
- ## RECK 0 0 1
- ## blood vessel development tissue morphogenesis cell adhesion
- ## PTK2 1 0 0
- ## GNA13 1 0 0
- ## LEPR 1 0 0
- ## APOE 1 0 0
- ## CXCR4 1 0 0
- ## RECK 1 0 0
- ## plasma membrane logFC
- ## PTK2 1 -0.6527904
- ## GNA13 1 0.3711599
- ## LEPR 1 2.6539788
- ## APOE 1 0.8698346
- ## CXCR4 1 -2.5647537
- ## RECK 1 3.6926860
Dans l'exemple, nous avons passé deux paramètres. Si seul le paramètre gènes est spécifié, le résultat est une liste de gènes sélectionnés et de toutes les constructions de processus avec au moins un gène spécifié.0-1matrice ; si seulement spécifiéprocessparamètres, le résultat est que tous les gènes génèrent0-1 Matrice de gènes affectés à au moins un processus de la liste. Sachez que la spécification uniquement des gènes et des paramètres de processus peut entraîner une très grande matrice 0-1, ce qui entraînerait des résultats de visualisation confus.
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
-
- # Generate the matrix with a list of selected genes
- chord_genes <- chord_dat(data = circ, genes = EC$genes)
- head(chord_genes)
-
- ## heart development vasculature development blood vessel development
- ## PTK2 0 1 1
- ## GNA13 0 1 1
- ## LEPR 0 1 1
- ## APOE 0 1 1
- ## CXCR4 0 1 1
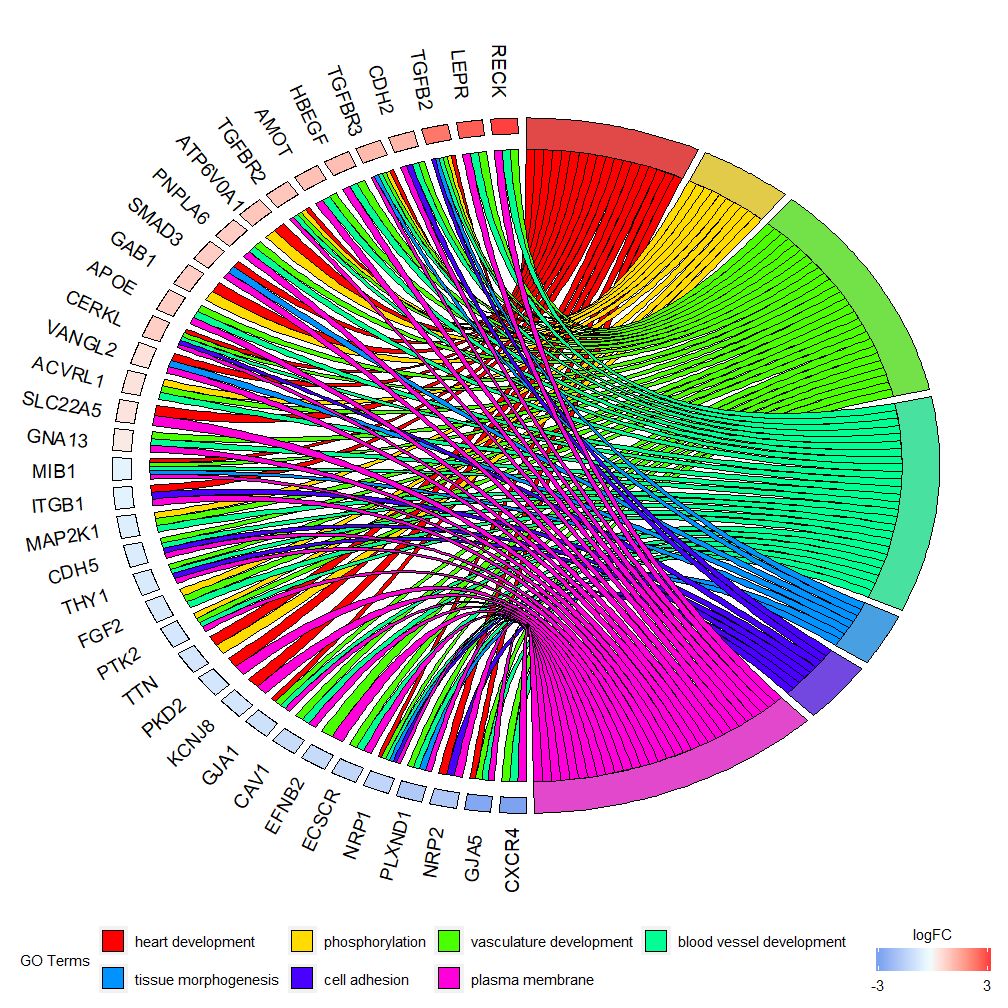
Ce graphique est destiné à montrer un sous-ensemble plus petit de données de grande dimension. Il y a principalement deux paramètres réglables :gene.orderetnlfc . Le paramètre gènes peut être spécifié comme « logFC », « alphabétique », « aucun ». En fait, nous spécifions généralement le paramètre des gènes comme logFC ; le paramètre nlfc est l'un des paramètres les plus importants de cette fonction, car il peut gérer la façon dont chaque gène a 0 ou plusieurs valeurs logFC présentées dans la matrice. Nous devons donc spécifier des paramètres pour éviter les erreurs.
Par exemple, si vous avez une matrice sans valeurs logFC, vous devez définirnlfc=0 ; Ou effectuez une analyse d'expression différentielle sur des gènes dans plusieurs conditions ou lots. Dans ce cas, chaque gène contient plusieurs valeurs logFC et le numéro de colonne nlfc=logFC doit être défini. La valeur par défaut est « 1 » car on pense que la plupart du temps, il n'y aura qu'une seule valeur logFC par gène. Utilisez le paramètre space pour définir l'espace entre les rectangles colorés représentant logFC. Le paramètre gene.size spécifie la taille de la police du nom du gène et gene.space spécifie la taille de l'espace entre les noms de gènes.
- chord <- chord_dat(data = circ, genes = EC$genes, process = EC$process)
- GOChord(chord, space = 0.02, gene.order = 'logFC', gene.space = 0.25, gene.size = 5)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
Peut être défini en fonction de la valeur logFCgene.order=‘logFC’ , triez les gènes en fonction de leurs valeurs logFC. Parfois, l'image peut devenir un peu encombrée et cela peut être automatisé en utilisant le paramètre limit pour réduire le nombre de gènes ou de voies affichés. La limite est un vecteur avec deux valeurs seuil (la valeur par défaut est c(0,0)). La première valeur précise le nombre minimum de voies auxquelles le gène doit être attribué. La deuxième valeur détermine le nombre de gènes attribués à la voie.
- # 仅显示分配给至少三个通路的基因
- GOChord(chord, limit = c(3, 0), gene.order = 'logFC')
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
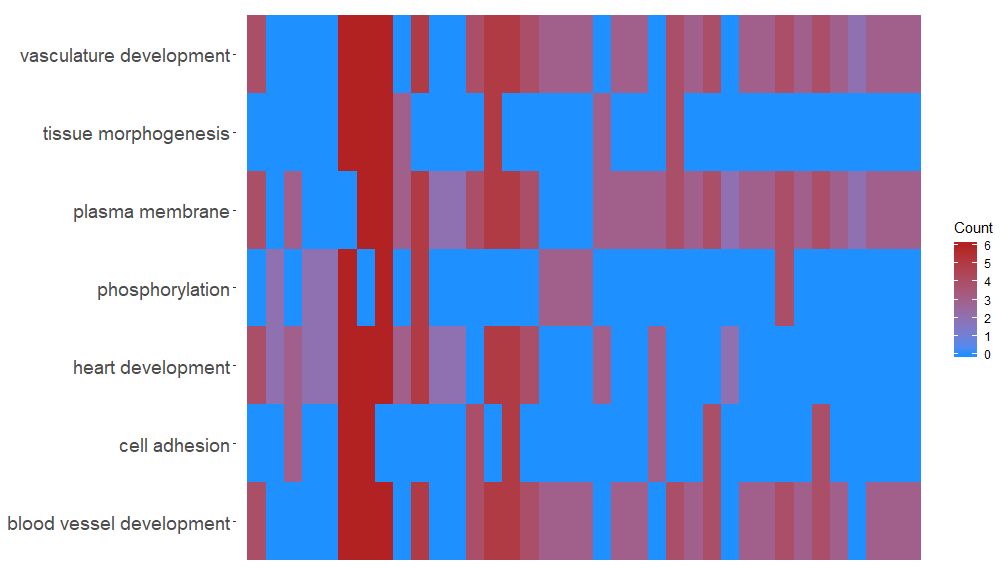
La fonction GOHeat peut afficher la relation entre les gènes et les voies à l'aide d'une carte thermique, similaire à GOChord. Les processus biologiques sont affichés horizontalement et les gènes sont affichés verticalement. Chaque colonne est divisée en petits rectangles et la couleur dépend généralement de la valeur logFC. De plus, les gènes enrichis dans des voies fonctionnelles similaires ont été regroupés. Il existe deux modes de sélection des couleurs de la carte thermique, en fonction des paramètres nlfc. Si nlfc = 0, la couleur est le nombre de voies enrichies pour chaque gène. Voir les exemples pour plus de détails :
- # First, we use the chord object without logFC column to create the heatmap
- GOHeat(chord[,-8], nlfc = 0)
GOHeat(chord[,-8])La couleur correspond au logFC du gène dans le cas nlfc = 1
GOHeat(chord, nlfc = 1, fill.col = c('red', 'yellow', 'green'))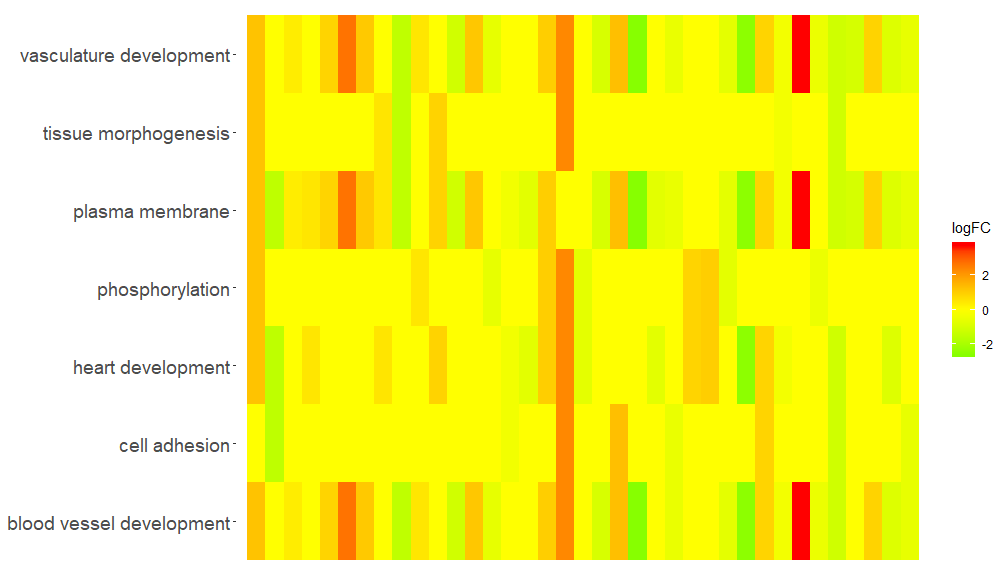
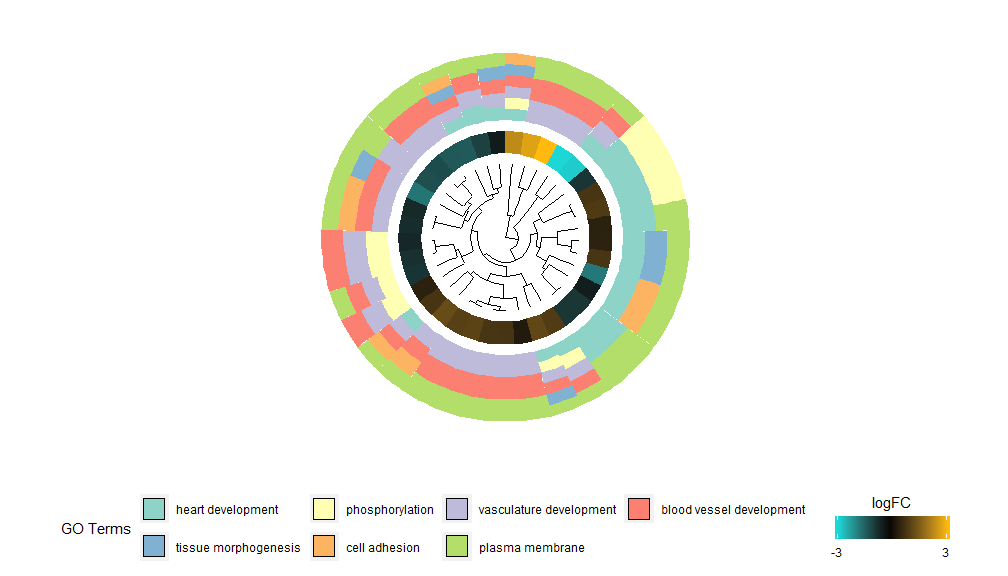
L'idée derrière la fonctionnalité GOCluster est d'afficher autant d'informations que possible. Voici un exemple:
- GOCluster(circ, EC$process, clust.by = 'logFC', term.width = 2)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
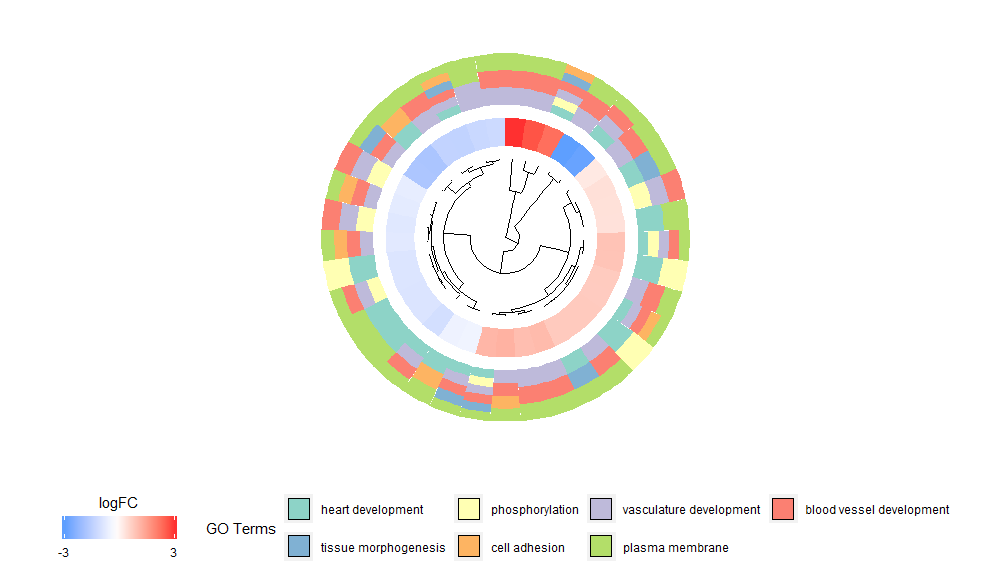
Le regroupement hiérarchique est une méthode d'analyse de regroupement non supervisée populaire pour l'expression des gènes qui garantit un regroupement impartial des gènes par modèle d'expression, de sorte que les grappes regroupées peuvent contenir plusieurs groupes de gènes co-régulés ou fonctionnellement liés. GOCluster utilise lehclust La méthode effectue un regroupement hiérarchique de profils d'expression génique. Si vous souhaitez modifier la métrique de distance ou l'algorithme de clustering, utilisez respectivement les paramètres metric et cluster. Le dendrogramme résultant peut être converti à l'aide de ggdendro et visualisé avec ggplot2. Choisissez une disposition circulaire car elle est non seulement efficace mais aussi visuellement attrayante. Le premier cercle à côté du dendrogramme représente le logFC du gène, qui est en fait la feuille de l'arbre de regroupement. Si vous êtes intéressé par plusieurs contrastes vous pouvez modifier le paramètre nlfc, par défaut il est réglé sur "1" donc un seul anneau est dessiné. Les valeurs logFC sont codées par couleur à l'aide d'une échelle de couleurs définissable par l'utilisateur (lfc.col) ; le cercle suivant représente la voie attribuée au gène. Afin de bien paraître, le nombre de canaux a été réduit et la couleur des canaux peut être modifiée à l'aide du paramètre term.col.toujours disponible?GOCluster pour voir comment modifier les paramètres. Le paramètre le plus important de cette fonction est cluster.by, qui peut être spécifié pour être regroupé par modèles d'expression génique (« logFC », comme indiqué ci-dessus) ou par catégories fonctionnelles (« termes »).
- GOCluster(circ, EC$process, clust.by = 'term', lfc.col = c('darkgoldenrod1', 'black', 'cyan1'))
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
Les diagrammes de Venn peuvent être utilisés pour détecter les relations entre diverses listes de gènes exprimés différentiellement ou pour explorer l'intersection de plusieurs gènes de voies dans des analyses fonctionnelles. Les diagrammes de Venn montrent non seulement le nombre de gènes qui se chevauchent, mais également des informations sur le modèle d'expression du gène (généralement régulé positivement, souvent régulé négativement ou contre-régulé). Actuellement, jusqu'à trois ensembles de données sont utilisés en entrée. Les données d'entrée contiennent au moins deux colonnes : une pour les noms de gènes et une pour les valeurs logFC.
- l1 <- subset(circ, term == 'heart development', c(genes,logFC))
- l2 <- subset(circ, term == 'plasma membrane', c(genes,logFC))
- l3 <- subset(circ, term == 'tissue morphogenesis', c(genes,logFC))
- GOVenn(l1,l2,l3, label = c('heart development', 'plasma membrane', 'tissue morphogenesis'))
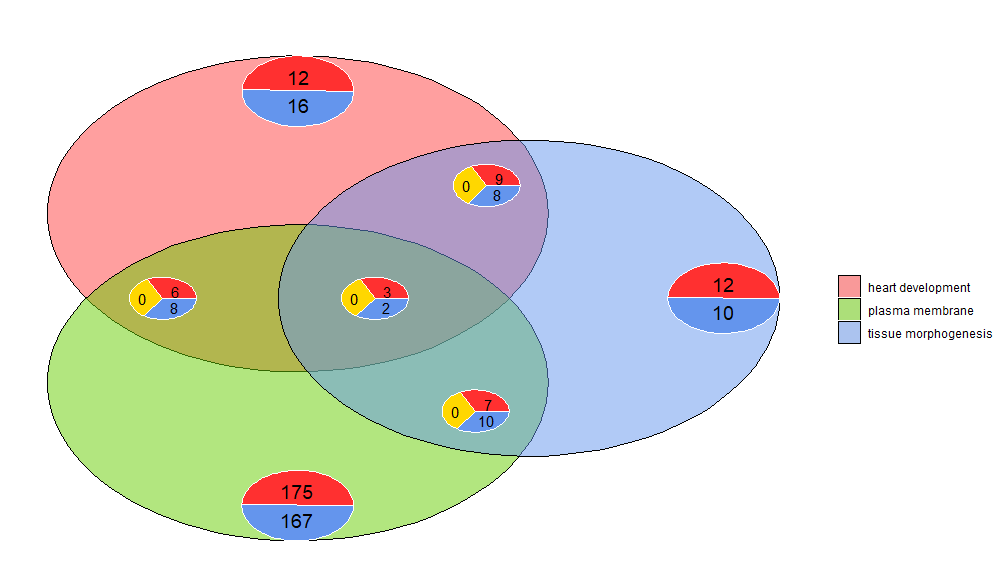
Par exemple, le développement cardiaque et la morphogenèse des tissus comportent 22 gènes, 12 sont régulés positivement et 10 sont régulés négativement. Il est important de noter que les diagrammes circulaires n’affichent pas d’informations redondantes. Par conséquent, si trois ensembles de données sont comparés, les gènes communs à tous les ensembles de données (le diagramme circulaire du milieu) ne sont pas inclus dans les autres diagrammes circulaires. Cet outil est disponible sur Shinyapp https://wwalter.shinyapps.io/Venn/, l'outil web est plus interactif, le cercle a une surface proportionnelle au nombre de gènes dans l'ensemble de données, et le curseur peut être utilisé pour déplacer le petit diagramme circulaire, et GOVenn propose toutes les options pour modifier la disposition de l'intrigue et également pour télécharger des images et des listes de gènes.
Page d'accueil du logiciel : https://wencke.github.io/

Il se consacre à la recherche technologique depuis plus de 30 ans et maîtrise divers langages tels que java, linux, javascript, php, css, etc. Il a apporté de nombreuses contributions dans le domaine de l'open source. station de documentation pour les développeurs pour partager certains problèmes de développement technologique pour référence future.

