
моя контактная информация
Почтамезофия@protonmail.com
2024-07-12
한어Русский языкEnglishFrançaisIndonesianSanskrit日本語DeutschPortuguêsΕλληνικάespañolItalianoSuomalainenLatina
Пакет GOPlot используется для визуализации биологических данных. Скорее, этот пакет интегрирует и визуализирует данные экспрессии с результатами функционального анализа.Но будь остороженЭтот пакет нельзя использовать для выполнения такого анализа, он предназначен только для визуализации результатов. . Во всех областях науки трудно реалистично описывать вещи из-за нехватки места и простоты, необходимой для получения результатов, поэтому информацию необходимо визуализировать и использовать изображения для ее передачи. Хорошо продуманная графика предоставляет больше информации на меньшем пространстве. Идея пакета заключается в том, чтобы дать пользователям возможность быстро изучать большие объемы данных, выявлять тенденции в данных и находить закономерности и корреляции в данных.
Визуализация данных может помочь нам найти ответы на биологические вопросы, оценить определенную гипотезу и даже открыть разные точки зрения для исследования различных проблем. А функции построения графиков этого пакета разработаны на основе иерархической структуры данных, начиная с общих данных и заканчивая подмножеством выбранных генов и соответствующих путей.
Поясним это конкретно на примере.
Мы вызываем данные, которые поступают с GOplot, который поступает из GEOGSE47067, содержащий транскриптомную информацию эндотелиальных клеток из двух тканей (мозга и сердца). Дополнительную информацию см. в статье Nolan et al. https://www.ncbi.nlm.nih.gov/pubmed/23871589, а затемДанные нормализуются и обнаруживаются дифференциально экспрессируемые гены., а затем используйте инструмент аннотаций функций DAVID (данные аннотаций DAVID обновляются медленно и сейчас не рекомендуются. Рекомендуется использоватьGo East, лучший онлайн-инструмент для анализа обогащения GOиЭтот веб-сайт, который может выполнить анализ обогащения всего за один шаг, до его публикации был процитирован более 350 раз CNS и другими организациями.Провести анализ обогащения,Освойте GSEA в одной статье, суперподробное руководство) Генная аннотация дифференциально экспрессируемых генов (adjusted p-value < 0.05 ) и анализ функционального обогащения. Этот набор данных содержит следующие пять категорий данных:
| имя | описывать | Размер набора данных |
|---|---|---|
| EC$eset | Нормализованная экспрессия генов в эндотелиальных клетках головного мозга и сердца (3 повтора) | 20644 х 7 |
| EC$genelist | Дифференциально экспрессируемые гены (скорректированное значение p <0,05) | 2039 х 7 |
| EC$david | Результаты анализа функционального обогащения дифференциальных генов с использованием DAVID | 174 х 5 |
| EC$ген | Гены и logFC | 37 х 2 |
| EC$процесс | Отобранные векторы признаков для обогащенных биологических процессов | 7 |
Мы хотим увидеть обогащенные GO пути дифференциально экспрессируемых генов, но прежде чем мы начнем рисовать, нам необходимо предоставить данные, соответствующие требованиям формата.Вообще говоря, данные, необходимые для построения графика, предоставляете вы сами, ноВ этом пакете есть функцияcircle_datМожет помочь нам разобраться с форматом данных。circle_datОн может комбинировать результаты анализа функционального обогащения выбранных генов и их значения logFC, в основном для дифференциально экспрессируемых генов.circle_dat Использование очень простое, просто прочитайте два данных. Первые данные содержат результаты анализа функционального обогащения, по крайней мере, с четырьмя столбцами (категория анализа функционального обогащения, путь, ген, скорректированное значение p).Вторые данные относятся к выбранному гену и его logFC, эти данные могут быть источником.limmaРезультаты статистического анализа (Примечание из биографий: Обязательно обратите внимание на два файлаКак называются геныБудьте последовательны, как и всеGene symbol ). Давайте рассмотрим упомянутые выше форматы данных на примерах.
- #安装已发布的稳定版本
- #install.packages('GOplot')
- #安装github上的开发版本
- #install_github('wencke/wencke.github.io')
- #载入包
- library(GOplot)
- #读入包内自带的数据
- data(EC)
- #查看功能富集分析结果的数据格式
- head(EC$david)
-
- ## Category ID Term
- ## 1 BP GO:0007507 heart development
- ## 2 BP GO:0001944 vasculature development
- ## 3 BP GO:0001568 blood vessel development
- ## 4 BP GO:0048729 tissue morphogenesis
- ## 5 BP GO:0048514 blood vessel morphogenesis
- ## 6 BP GO:0051336 regulation of hydrolase activity
- ## Genes
- ## 1 DLC1, NRP2, NRP1, EDN1, PDLIM3, GJA1, TTN, GJA5, ZIC3, TGFB2, CERKL, GATA6, COL4A3BP, GAB1, SEMA3C, MKL2, SLC22A5, MB, PTPRJ, RXRA, VANGL2, MYH6, TNNT2, HHEX, MURC, MIB1, FOXC2, FOXC1, ADAM19, MYL2, TCAP, EGLN1, SOX9, ITGB1, CHD7, HEXIM1, PKD2, NFATC4, PCSK5, ACTC1, TGFBR2, NF1, HSPG2, SMAD3, TBX1, TNNI3, CSRP3, FOXP1, KCNJ8, PLN, TSC2, ATP6V0A1, TGFBR3, HDAC9
- ## 2 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, EFNB2, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 3 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 4 DLC1, ENAH, NRP1, PGF, ZIC2, TGFB2, CD44, ILK, SEMA3C, RET, AR, RXRA, VANGL2, LEF1, TNNT2, HHEX, MIB1, NCOA3, FOXC2, FOXC1, TGFB1I1, WNT5A, COBL, BBS4, FGFR3, TNC, BMPR2, CTNND1, EGLN1, NR3C1, SOX9, TCF7L1, IGF1R, FOXQ1, MACF1, HOXA5, BCL2, PLXND1, CAR2, ACTC1, TBX4, SMAD3, FZD3, SHANK3, FZD6, HOXB4, FREM2, TSC2, ZIC5, TGFBR3, APAF1
- ## 5 GNA13, CAV1, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, CDH2, MEIS1, WT1, TGFB2, WARS, PTK2, CERKL, APOE, CXCR4, ANG, SEMA3C, PLCD1, NOS2, MKL2, PLXND1, FIGF, FGF2, PTPRJ, TGFBR2, TBX4, NF1, TBX1, TNNI3, PNPLA6, VASH1, THY1, NUS1, MEOX2, ECSCR, AMOT, HBEGF, FOXC2, FOXC1, HDAC7
- ## 6 CAV1, XIAP, AGFG1, ADORA2A, TNNC1, TBC1D9, LEPR, ABHD5, EDN1, ASAP2, ASAP3, SMAP1, TBC1D12, ANG, TBC1D14, MTCH1, TBC1D13, TBC1D4, TBC1D30, DHCR24, HIP1, VAV3, NOS1, NF1, MYH6, RICTOR, TBC1D22A, THY1, PLCE1, RNF7, NDEL1, CHML, IFT57, ACAP2, TSC2, ERN1, APAF1, ARAP3, ARAP2, ARAP1, HTR2A, F2R
- ## adj_pval
- ## 1 0.000002170
- ## 2 0.000010400
- ## 3 0.000007620
- ## 4 0.000119000
- ## 5 0.000720000
- ## 6 0.001171166
-
- #查看基因的数据格式
- head(EC$genelist)
-
- ## ID logFC AveExpr t P.Value adj.P.Val B
- ## 1 Slco1a4 6.645388 1.2168670 88.65515 1.32e-18 2.73e-14 29.02715
- ## 2 Slc19a3 6.281525 1.1600468 69.95094 2.41e-17 2.49e-13 27.62917
- ## 3 Ddc 4.483338 0.8365231 65.57836 5.31e-17 3.65e-13 27.18476
- ## 4 Slco1c1 6.469384 1.3558865 59.87613 1.62e-16 8.34e-13 26.51242
- ## 5 Sema3c 5.515630 2.3252117 58.53141 2.14e-16 8.81e-13 26.33626
- ## 6 Slc38a3 4.761755 0.9218670 54.11559 5.58e-16 1.76e-12 25.70308
Поняв два формата входных данных, вы можете использоватьcirlce_datфункция для генерации данных чертежа.
- # 生成画图所需的数据格式
- circ <- circle_dat(EC$david, EC$genelist)
-
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
circОбъект имеет восемь столбцов данных, а именно
категория: BP (биологический процесс), CC (клеточный компонент) или MF (молекулярная функция)
ID: идентификатор GO (необязательный столбец; если вы хотите использовать инструмент функционального анализа, не основанный на идентификаторе GO, вы не можете выбрать столбец ID; идентификатор здесь также может быть идентификатором KEGG)
термин: путь GO
подсчет: количество генов в каждом пути
ген: имя гена - logFC: значение logFC каждого гена
adj_pval: скорректированное значение p, пути с adj_pval<0,05 считаются значительно обогащенными.
zscore: zscore не относится к методу статистической нормализации, но представляет собой легко вычисляемое значение, позволяющее оценить, с большей вероятностью снижается биологический процесс (/молекулярная функция/клеточный компонент) (отрицательное значение) или увеличивается (положительное значение).Метод расчета представляет собой количество генов с повышенной регуляцией минус количество генов с пониженной регуляцией, разделенное на квадратный корень из числа генов в каждом пути.
Когда мы впервые смотрим на данные, мы хотим показать на графике как можно больше путей, а также мы хотим найти ценные пути, поэтому нам нужны некоторые параметры для оценки важности. Гистограммы часто используются для описания выборочных данных, поэтому мы можем использовать функцию GOBar, чтобы быстро создать красивую гистограмму.
Сначала создается простая гистограмма. Горизонтальная ось.GO Terms, по их словамzscoreСортировка столбцов по вертикальной оси;-log(adj p-value);Цвет представляетzscore, синий указываетz-scoreявляется отрицательным значением, экспрессия гена в соответствующем пути с большей вероятностью снизится (обозначено красным).z-score является положительным значением, то экспрессия гена в соответствующем пути с большей вероятностью увеличится. При желании порядок можно изменить, установив для параметра order.by.zscore значение FALSE, в этом случае бары упорядочиваются в зависимости от их значимости.
- # 生成简单的条形图
- GOBar(subset(circ, category == 'BP'))
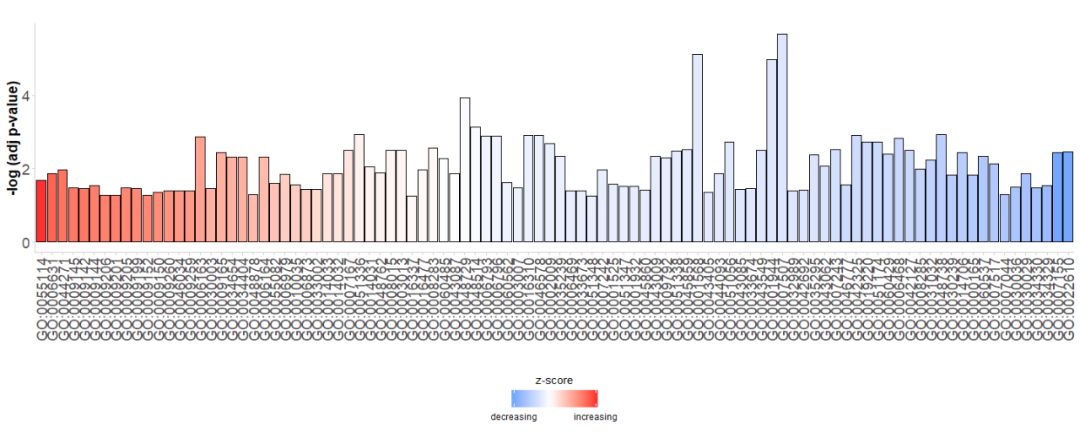
#GOBar(subset(circ, category == 'BP',order.by.zscore=FALSE))Кроме того, измените параметр отображения, чтобы построить гистограмму в соответствии с категорией канала.
- #根据通路的类别来绘制条形图
- GOBar(circ, display = 'multiple')
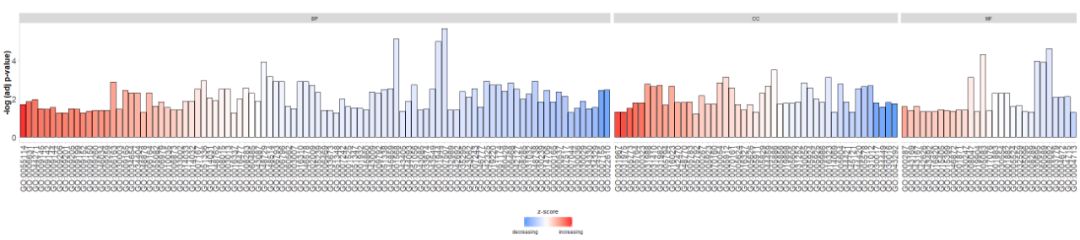
Добавьте заголовок и используйте параметрыzsc.colИзменятьzscoreцвет.
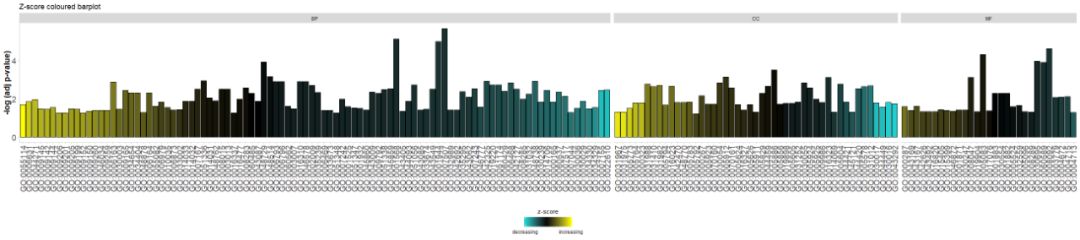
- # Facet the barplot, add a title and change the colour scale for the z-score
- GOBar(circ, display = 'multiple', title = 'Z-score coloured barplot', zsc.col = c('yellow', 'black', 'cyan'))
Гистограммы очень распространены и просты для понимания, но мы можем использовать пузырьковые диаграммы для отображения дополнительной информации о данных.
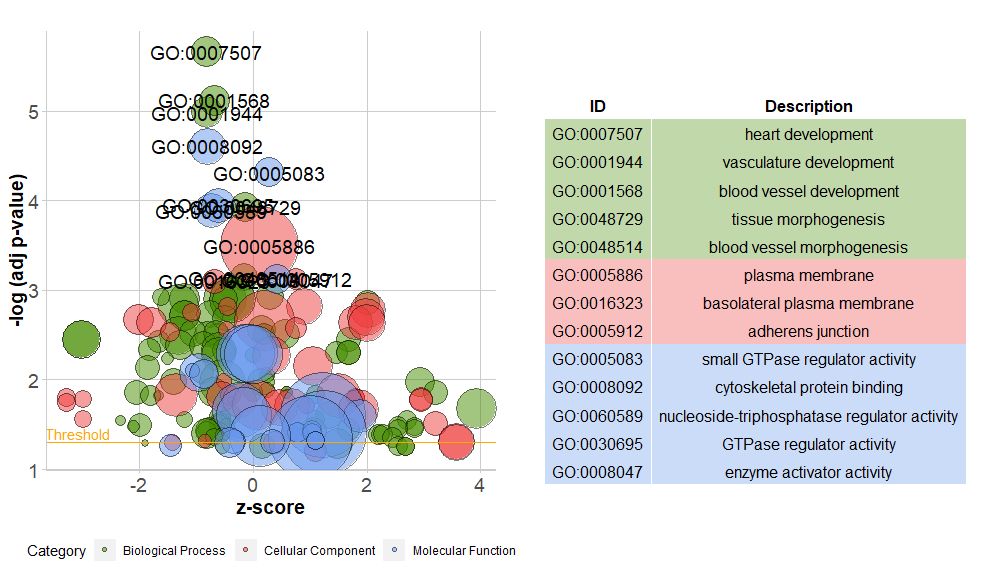
Горизонтальная осьzscore;Вертикальная ось-log(adj p-value), аналогично гистограмме, чем она выше, тем значительнее обогащение связано с количеством генов в соответствующем пути (;circ$count ); цвет соответствует категории, соответствующей пути, зеленый — биологический процесс, красный — клеточный компонент, синий — молекулярная функция.Может быть введен по?GOBubble См. страницу справки функции GOBubble, чтобы изменить все параметры изображения. По умолчанию каждый круг отмечен соответствующим идентификатором GO, а справа также отображается таблица, показывающая соответствующую взаимосвязь между идентификатором GO и термином GO.Параметры могут быть установлены с помощьюtable.legendдляFALSE чтобы скрыть это. Если вы хотите отобразить описание пути, установите для идентификатора параметра значение FALSE.Однако из-за ограниченного пространства и перекрывающихся кругов отмечены не все круги, а только-log(adj p-value) > 3(по умолчанию 5).
- # 生成泡泡图,并展示-log(adj p-value) > 3 的通路的GO ID
- GOBubble(circ, labels = 3)
Если вы хотите добавить заголовок к пузырьковой диаграмме или указать цвет кружка и отобразить пути каждой категории отдельно, а также изменить отображаемый порог идентификатора GO ID, вы можете добавить следующие параметры:
GOBubble(circ, title = 'Bubble plot', colour = c('orange', 'darkred', 'gold'), display = 'multiple', labels = 3)
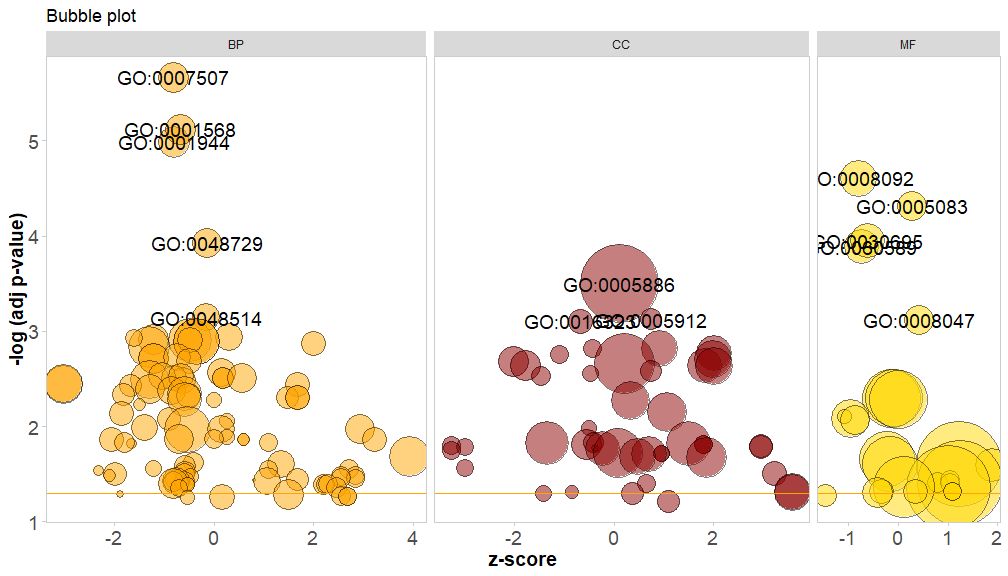
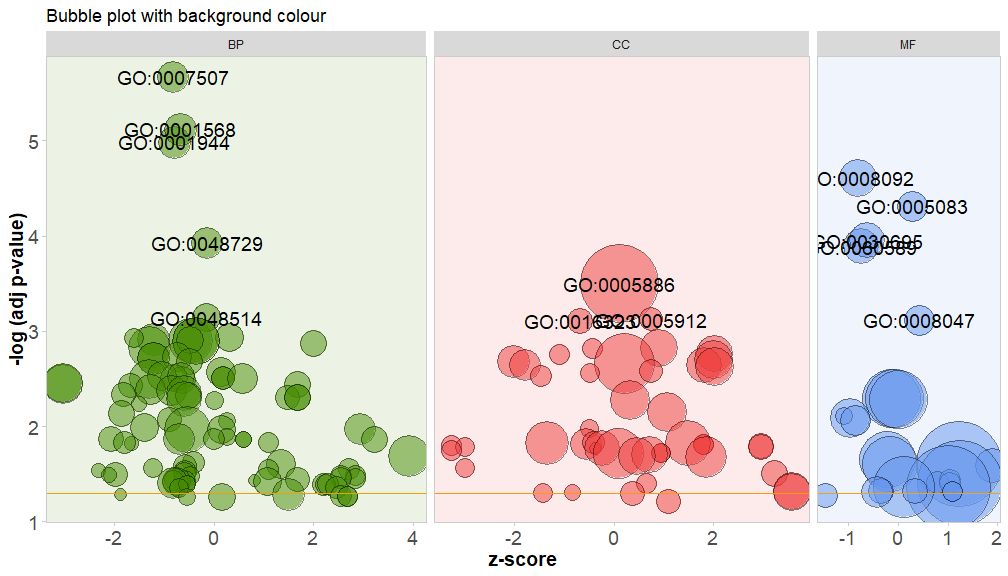
Раскрасьте фон класса канала, установив для параметра bg.col значение TRUE.
GOBubble(circ, title = 'Bubble plot with background colour', display = 'multiple', bg.col = T, labels = 3)
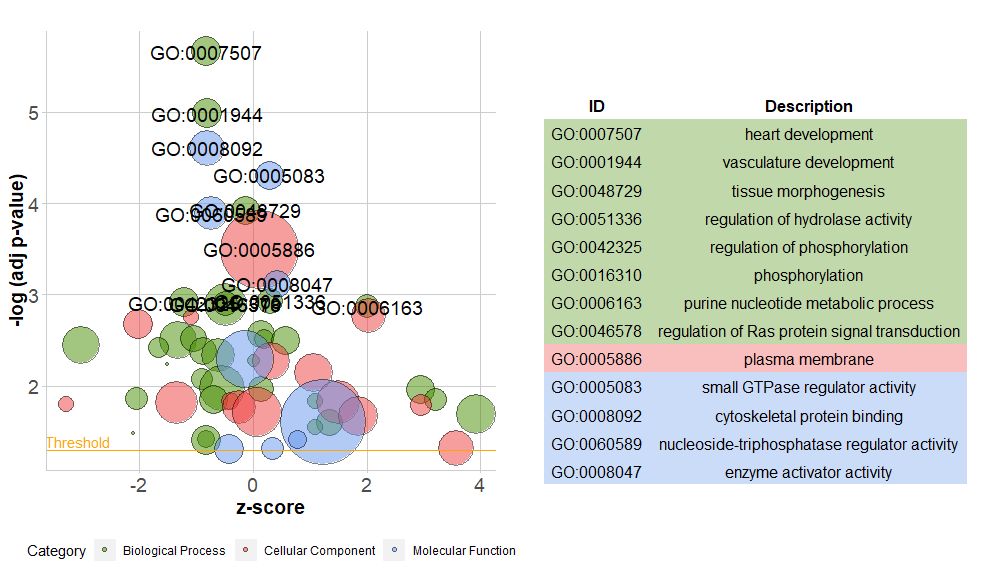
Новая версия пакета содержит новую функциюreduce_overlap , эта функция может уменьшить количество избыточных элементов, то есть она может удалить все пути, перекрытие генов которых больше или равно установленному порогу, и сохранить только один путь из каждой группы в качестве репрезентативного, независимо от отображения всех пути в ГО. За счет уменьшения количества избыточных терминов значительно улучшается читабельность графиков (например, пузырьковых).
- # reduce_overlap,参数设置为0.75
- reduced_circ <- reduce_overlap(circ, overlap = 0.75)
-
- GOBubble(reduced_circ, labels = 2.8)
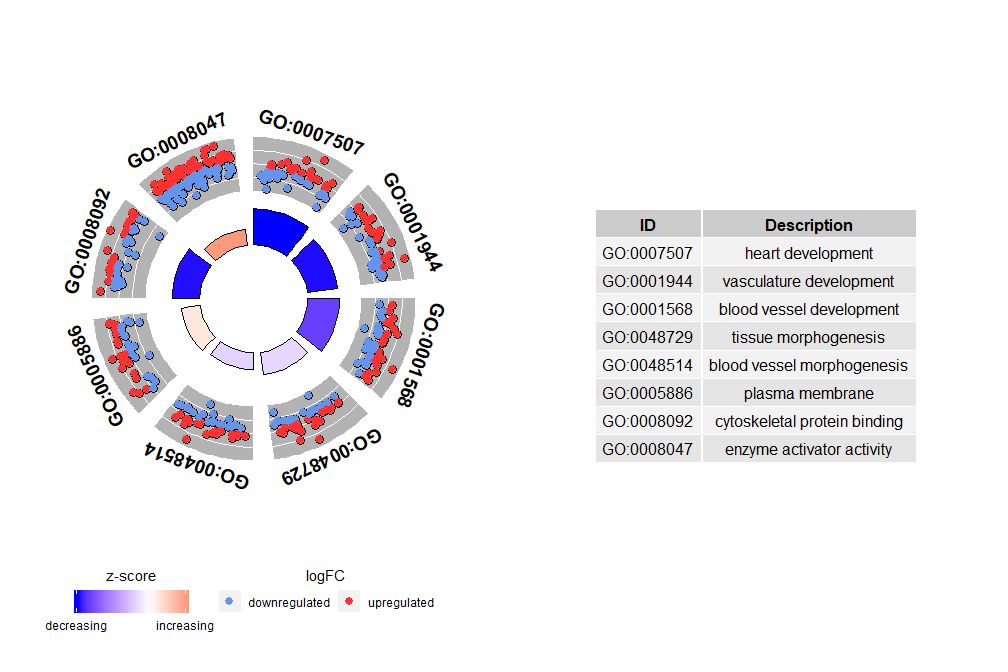
Хотя график, показывающий всю информацию, может помочь нам определить, какие пути являются наиболее значимыми, реальность по-прежнему зависит от гипотез и идей, которые вы хотите подтвердить с помощью данных, и наиболее важные пути не обязательно могут быть теми, которые вас интересуют. Таким образом, при ручном выборе ценного набора путей (EC$process ), нам нужна диаграмма, которая покажет нам более подробную информацию об этом конкретном наборе путей.Но с представлением этих цифр возникает проблема: иногда их трудно интерпретировать.zscore Информация предоставлена.В конце концов, этот метод расчета не является универсальным. Как показано выше, это просто количество генов с повышающей регуляцией минус количество генов с пониженной регуляцией, деленное на квадратный корень из числа генов в каждом пути, используя метод.GOCircleПолученный график также подчеркивает этот факт.
Внешний круг круговой диаграммы показывает значение logFC генов каждого пути в виде разбросанных точек. Красные круги указывают на усиление, а синие — на понижение.Параметры можно использоватьlfc.col Изменить цвет. Это также объясняет, почему в некоторых случаях очень важные пути имеют z-показатели, близкие к нулю. Нулевой zscore не означает, что канал неважен. Это просто показывает, что zscore является грубой мерой, поскольку очевидно, что zscore также не учитывает функциональный уровень и активационную зависимость отдельных генов в биологических процессах.
GOCircle(circ)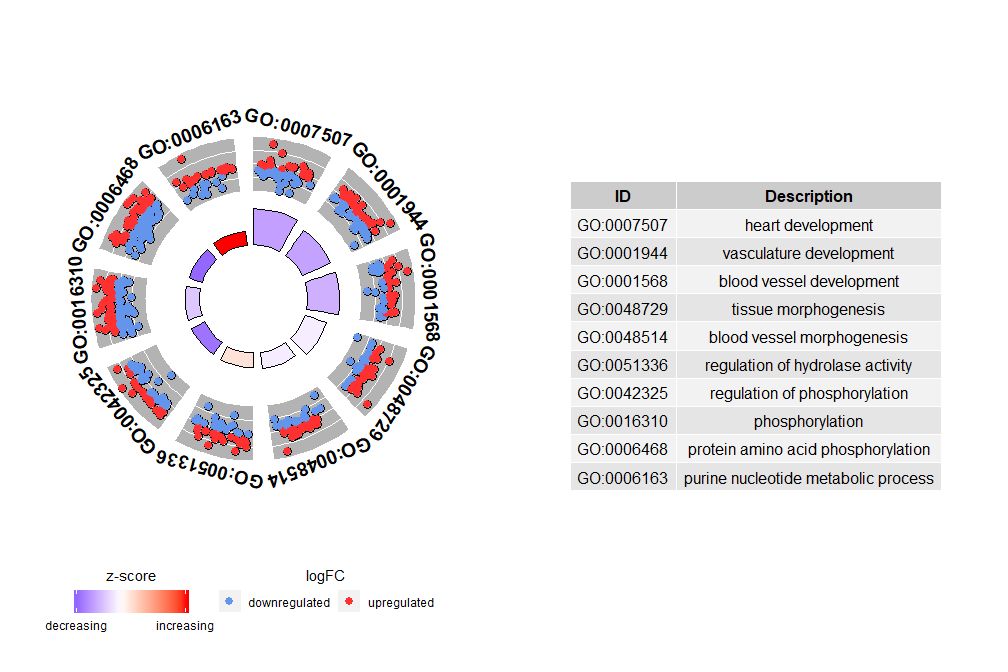
nsub Параметры могут быть заданными числами или векторами символов. Если это вектор символов, он содержит идентификатор GO или путь для отображения;
- # 生成特定通路的圈图
- IDs <- c('GO:0007507', 'GO:0001568', 'GO:0001944', 'GO:0048729', 'GO:0048514', 'GO:0005886', 'GO:0008092', 'GO:0008047')
- GOCircle(circ, nsub = IDs)
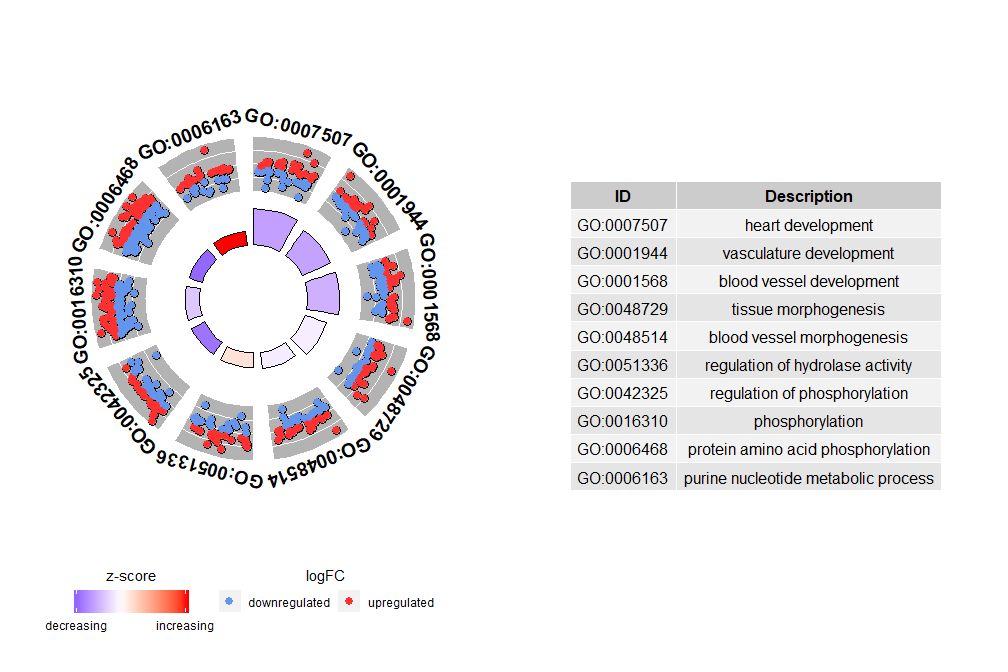
Если nsub — числовой вектор, число определяет отображаемый номер. Он начинается с первой строки входного фрейма данных. Эта визуализация работает только с меньшими данными. Максимальное количество каналов по умолчанию равно 12. Хотя количество каналов уменьшается, объем отображаемой информации увеличивается.
- # 圈图展示数据前十个通路
- GOCircle(circ, nsub = 10)
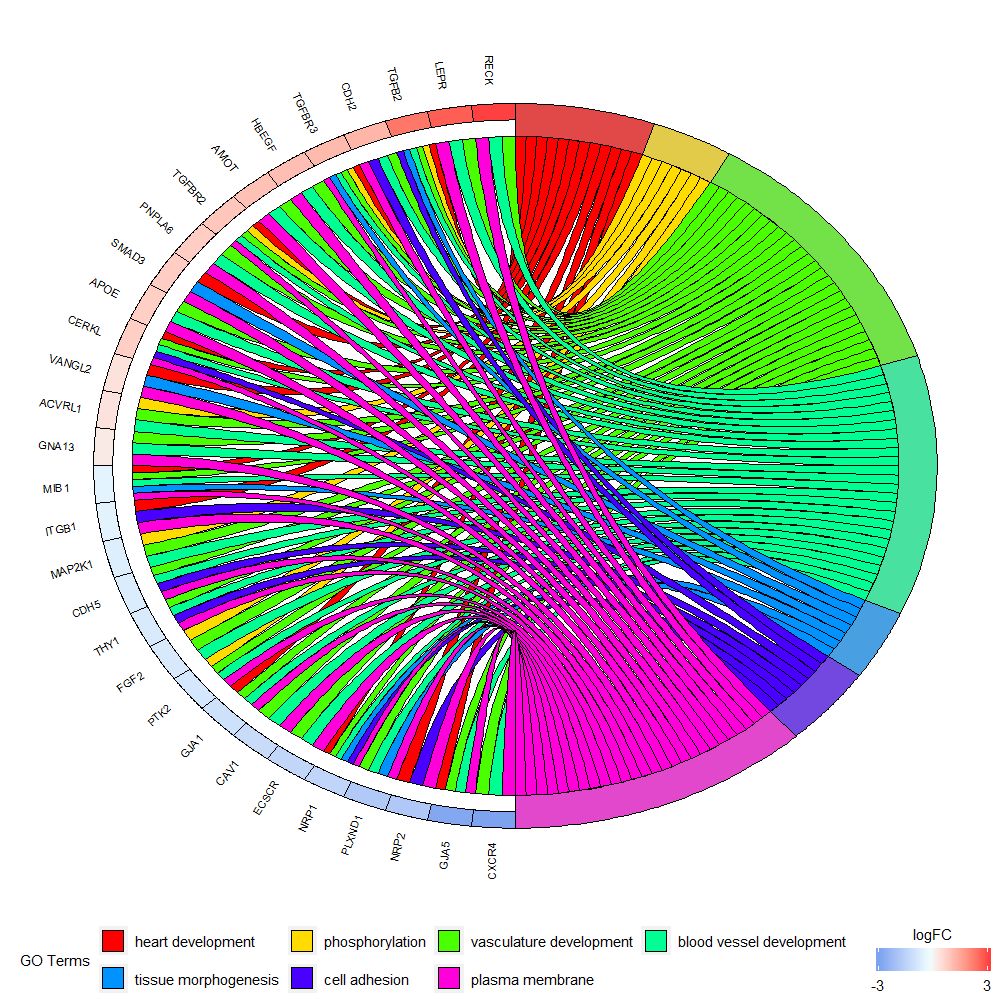
GOChord может отображать взаимосвязь между выбранными генами и путями, а также logFC генов.Сначала вам нужно ввести матрицу, которую вы можете построить самостоятельно.0-1Матрица, вы также можете использовать функцииchord_dat Построить. Эта функция имеет три параметра: данные, гены и процесс, из которых последние два параметра должны иметь хотя бы один параметр.Тогда функцияcircle_datОбъедините данные выражений с результатами функционального анализа.
Гистограммы и пузырьковые диаграммы могут дать вам первое представление о данных. Теперь вы можете выбрать некоторые гены и пути, которые, по нашему мнению, являются ценными. Хотя GOCircle добавляет слой для отображения значения экспрессии генов в путях, в нем отсутствует единая информация. о взаимоотношениях между генами и множественными путями. Нелегко выяснить, связаны ли определенные гены с несколькими процессами. GOChord компенсирует недостатки GOCircle. Строки сгенерированных данных — это гены, а столбцы — пути. «0» означает, что ген не присвоен пути, а «1» — наоборот.
- # 找到感兴趣的的基因,这里我们以EC$genes为例
- head(EC$genes)
-
- ## ID logFC
- ## 1 PTK2 -0.6527904
- ## 2 GNA13 0.3711599
- ## 3 LEPR 2.6539788
- ## 4 APOE 0.8698346
- ## 5 CXCR4 -2.5647537
- ## 6 RECK 3.6926860
-
- # 获得感兴趣基因的通路
- EC$process
-
- ## [1] "heart development" "phosphorylation"
- ## [3] "vasculature development" "blood vessel development"
- ## [5] "tissue morphogenesis" "cell adhesion"
- ## [7] "plasma membrane"
-
- # 使用chord_dat构建矩阵
- chord <- chord_dat(circ, EC$genes, EC$process)
- head(chord)
-
- ## heart development phosphorylation vasculature development
- ## PTK2 0 1 1
- ## GNA13 0 0 1
- ## LEPR 0 0 1
- ## APOE 0 0 1
- ## CXCR4 0 0 1
- ## RECK 0 0 1
- ## blood vessel development tissue morphogenesis cell adhesion
- ## PTK2 1 0 0
- ## GNA13 1 0 0
- ## LEPR 1 0 0
- ## APOE 1 0 0
- ## CXCR4 1 0 0
- ## RECK 1 0 0
- ## plasma membrane logFC
- ## PTK2 1 -0.6527904
- ## GNA13 1 0.3711599
- ## LEPR 1 2.6539788
- ## APOE 1 0.8698346
- ## CXCR4 1 -2.5647537
- ## RECK 1 3.6926860
В примере мы передали два параметра. Если указан только параметр генов, результатом будет список выбранных генов и всех конструкций процессов, в которых есть хотя бы один указанный ген.0-1матрица, если только указана;processпараметров, в результате все гены генерируют0-1 Матрица генов, присвоенных хотя бы одному процессу в списке. Имейте в виду, что указание только генов и параметров процесса может привести к очень большой матрице 0-1, что приведет к путанице в результатах визуализации.
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
-
- # Generate the matrix with a list of selected genes
- chord_genes <- chord_dat(data = circ, genes = EC$genes)
- head(chord_genes)
-
- ## heart development vasculature development blood vessel development
- ## PTK2 0 1 1
- ## GNA13 0 1 1
- ## LEPR 0 1 1
- ## APOE 0 1 1
- ## CXCR4 0 1 1
Эта диаграмма предназначена для отображения меньшего подмножества многомерных данных. В основном можно регулировать два параметра:gene.orderиnlfc . Параметр генов может быть указан как «logFC», «алфавитный», «нет». Фактически, мы обычно указываем параметр генов как logFC; параметр nlfc является одним из наиболее важных параметров этой функции, поскольку он может обрабатывать то, что каждый ген имеет 0 или более значений logFC, представленных в матрице. Поэтому нам следует указать параметры, чтобы избежать ошибок.
Например, если у вас есть матрица без значений logFC, вы должны установитьnlfc=0 ; Или выполнить дифференциальный анализ экспрессии генов в нескольких условиях или партиях. В этом случае каждый ген содержит несколько значений logFC, и необходимо установить номер столбца nlfc=logFC. Значением по умолчанию является «1», поскольку считается, что в большинстве случаев для каждого гена будет только одно значение logFC. Используйте параметр space, чтобы определить пространство между цветными прямоугольниками, представляющими logFC. Параметр Gene.size определяет размер шрифта имени гена, а Gene.space определяет размер пробела между именами генов.
- chord <- chord_dat(data = circ, genes = EC$genes, process = EC$process)
- GOChord(chord, space = 0.02, gene.order = 'logFC', gene.space = 0.25, gene.size = 5)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
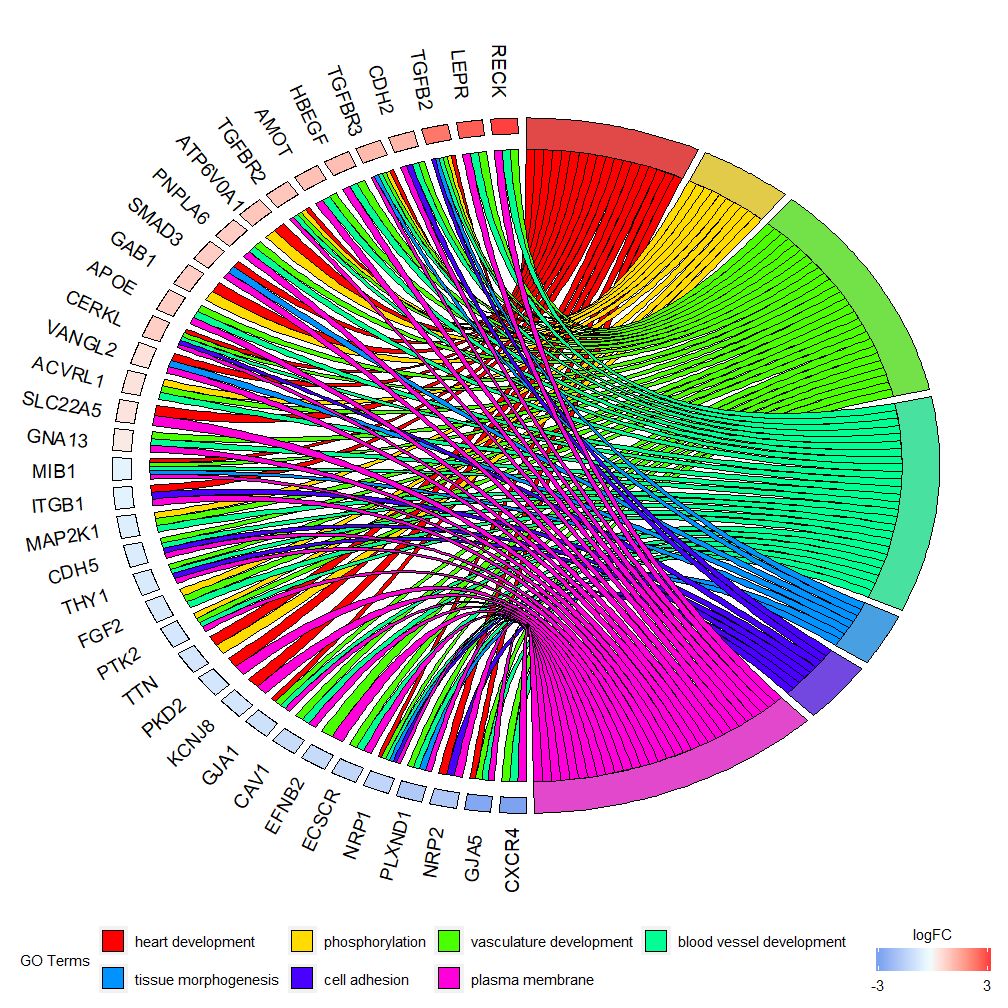
Может быть установлен в соответствии со значением logFC.gene.order=‘logFC’ гены сортируются в соответствии с их значениями logFC. Иногда изображение может стать немного перегруженным, и это можно автоматизировать, используя параметр limit, чтобы уменьшить количество отображаемых генов или путей. Limit — вектор с двумя значениями отсечки (по умолчанию — c(0,0)). Первое значение указывает минимальное количество путей, которым должен быть назначен ген. Второе значение определяет количество генов, назначенных этому пути.
- # 仅显示分配给至少三个通路的基因
- GOChord(chord, limit = c(3, 0), gene.order = 'logFC')
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
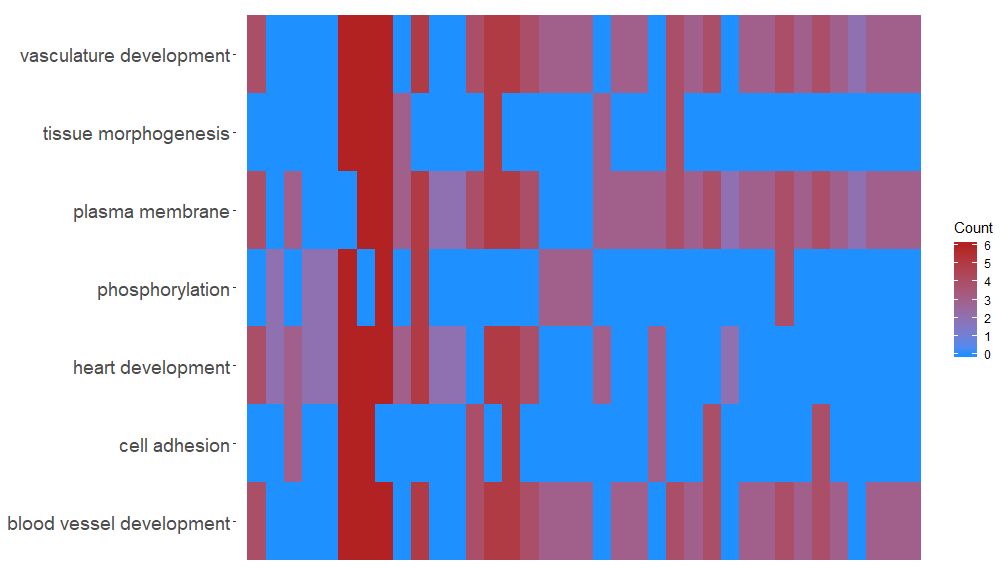
Функция GOHeat может отображать взаимосвязь между генами и путями с помощью тепловой карты, аналогичной GOChord. Биологические процессы отображаются горизонтально, а гены — вертикально. Каждый столбец разделен на небольшие прямоугольники, а цвет обычно зависит от значения logFC. Кроме того, были сгруппированы гены, имеющие схожие функциональные пути. Существует два режима выбора цвета тепловой карты, в зависимости от параметров nlfc. Если nlfc = 0, цвет обозначает количество обогащенных путей для каждого гена. Подробности смотрите в примерах:
- # First, we use the chord object without logFC column to create the heatmap
- GOHeat(chord[,-8], nlfc = 0)
GOHeat(chord[,-8])Цвет соответствует logFC гена в случае nlfc = 1.
GOHeat(chord, nlfc = 1, fill.col = c('red', 'yellow', 'green'))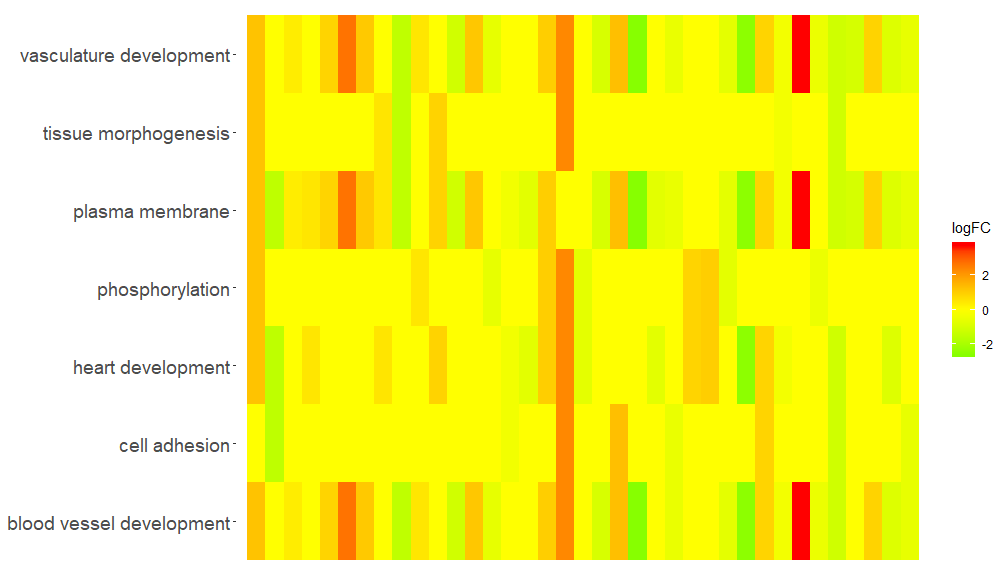
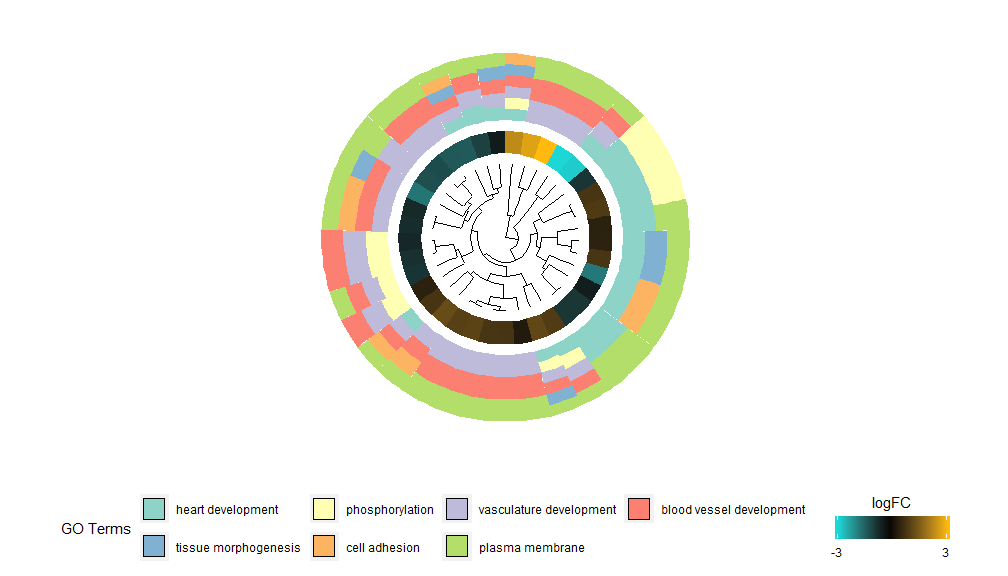
Идея функциональности GOCluster заключается в отображении как можно большего количества информации. Вот пример:
- GOCluster(circ, EC$process, clust.by = 'logFC', term.width = 2)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
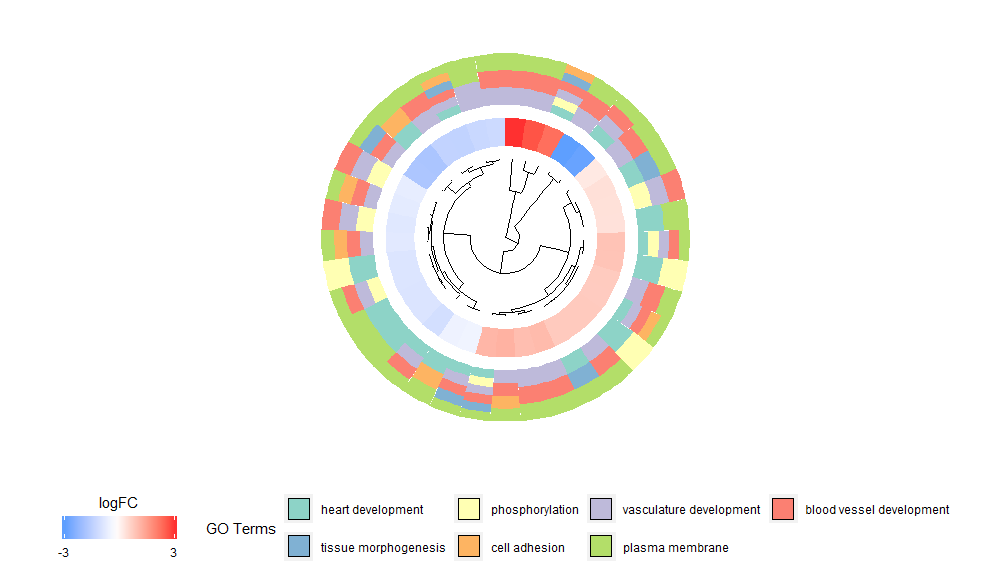
Иерархическая кластеризация — это популярный метод неконтролируемого кластерного анализа экспрессии генов, который обеспечивает объективную группировку генов по характеру экспрессии, так что кластеры могут содержать несколько групп совместно регулируемых или функционально связанных генов. GOCluster используетhclust Метод осуществляет иерархическую кластеризацию профилей экспрессии генов. Если вы хотите изменить метрику расстояния или алгоритм кластеризации, используйте параметры metric и clust соответственно. Полученную дендрограмму можно преобразовать с помощью ggdendro и визуализировать с помощью ggplot2. Выбирайте круглую планировку, поскольку она не только эффективна, но и визуально привлекательна. Первый кружок рядом с дендрограммой представляет logFC гена, который на самом деле является листом дерева кластеризации. Если вас интересуют несколько контрастов, вы можете изменить параметр nlfc, по умолчанию он установлен на «1», поэтому рисуется только одно кольцо. Значения logFC имеют цветовую маркировку с использованием определяемой пользователем цветовой шкалы (lfc.col); следующий кружок представляет путь, присвоенный гену. Чтобы выглядеть хорошо, количество каналов было уменьшено, а цвет каналов можно изменить с помощью параметра term.col.до сих пор доступен?GOCluster чтобы увидеть, как изменить параметры. Наиболее важным параметром этой функции являетсяcluster.by, который можно указать для кластеризации по шаблонам экспрессии генов («logFC», как показано выше) или функциональным категориям («термины»).
- GOCluster(circ, EC$process, clust.by = 'term', lfc.col = c('darkgoldenrod1', 'black', 'cyan1'))
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
Диаграммы Венна можно использовать для обнаружения взаимосвязей между различными списками дифференциально экспрессируемых генов или для изучения пересечения генов нескольких путей в функциональном анализе. Диаграммы Венна показывают не только количество перекрывающихся генов, но и информацию о характере экспрессии гена (обычно повышающая, часто понижающая или контррегулируемая). В настоящее время в качестве входных данных используется до трех наборов данных. Входные данные содержат как минимум два столбца: один для имен генов и один для значений logFC.
- l1 <- subset(circ, term == 'heart development', c(genes,logFC))
- l2 <- subset(circ, term == 'plasma membrane', c(genes,logFC))
- l3 <- subset(circ, term == 'tissue morphogenesis', c(genes,logFC))
- GOVenn(l1,l2,l3, label = c('heart development', 'plasma membrane', 'tissue morphogenesis'))
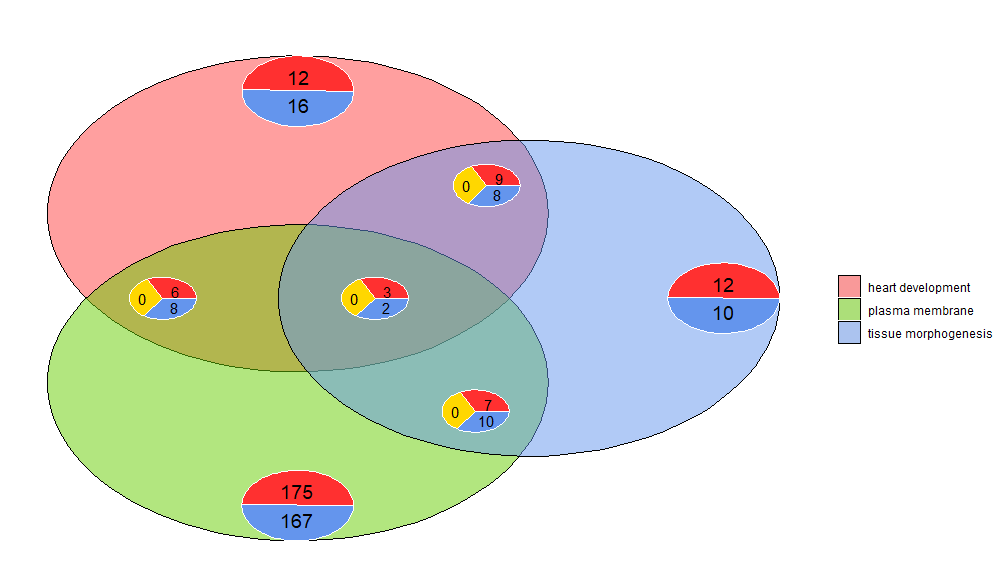
Например, развитие сердца и морфогенез тканей имеют 22 гена, 12 из которых имеют повышенную экспрессию, а 10 — отрицательную. Важно отметить, что круговые диаграммы не отображают избыточную информацию. Следовательно, если сравниваются три набора данных, гены, общие для всех наборов данных (средняя круговая диаграмма), не включаются в другие круговые диаграммы. Этот инструмент доступен в ShinyApp https://wwalter.shinyapps.io/Venn/, веб-инструмент более интерактивный, площадь круга пропорциональна количеству генов в наборе данных, а ползунок можно использовать для перемещения небольшая круговая диаграмма и имеет функции GOVenn со всеми опциями для изменения макета графика, а также для загрузки изображений и списков генов.
Домашняя страница программного обеспечения: https://wencke.github.io/

Он посвятил себя исследованию технологий более 30 лет и владеет различными языками, такими как Java, Linux, Javascript, php, css и т. д. Он внес большой вклад в область открытого исходного кода. Станция документации для разработчиков, где можно поделиться некоторыми проблемами в разработке технологий для дальнейшего использования. Все ознакомьтесь.

Почтамезофия@protonmail.com