
Mi información de contacto
Correo[email protected]
2024-07-12
한어Русский языкEnglishFrançaisIndonesianSanskrit日本語DeutschPortuguêsΕλληνικάespañolItalianoSuomalainenLatina
El paquete GOPlot se utiliza para la visualización de datos biológicos. Más bien, este paquete integra y visualiza datos de expresión con los resultados de análisis funcionales.Pero ten cuidadoEste paquete no se puede utilizar para realizar estos análisis, sólo para visualizar los resultados. . En todos los campos de la ciencia, es difícil describir cosas de manera realista debido a las limitaciones de espacio y la simplicidad requerida para los resultados, por lo que es necesario visualizar la información y utilizar imágenes para transmitirla. Los gráficos bien diseñados brindan más información en menos espacio. La idea del paquete es permitir a los usuarios examinar rápidamente grandes cantidades de datos, revelar tendencias en los datos y encontrar patrones y correlaciones en los datos.
La visualización de datos puede ayudarnos a encontrar respuestas a preguntas biológicas, juzgar una determinada hipótesis e incluso descubrir diferentes ángulos para investigar diferentes problemas. Y las funciones de trazado de este paquete se desarrollan en función de la estructura jerárquica de los datos, comenzando con los datos generales y terminando con un subconjunto de genes seleccionados y las vías correspondientes.
Expliquemoslo concretamente con un ejemplo.
A los datos que vienen con GOplot, que provienen de GEO, los llamamosGSE47067, que contiene información del transcriptoma de células endoteliales de dos tejidos (cerebro y corazón). Para obtener más información, consulte el artículo de Nolan et al. https://www.ncbi.nlm.nih.gov/pubmed/23871589.Los datos se normalizan y se encuentran genes expresados diferencialmente.y luego use la herramienta de anotación de funciones DAVID (los datos de anotación de DAVID se actualizan lentamente y no se recomiendan ahora. Se recomienda usarGo East, la mejor herramienta de análisis de enriquecimiento GO onlineyEste sitio web, que puede realizar análisis de enriquecimiento en un solo paso, ha sido citado más de 350 veces por CNS y otros antes de su publicación.Realizar análisis de enriquecimiento,Domina GSEA en un artículo, tutorial súper detallado) Anotación genética de genes expresados diferencialmente (adjusted p-value < 0.05 ) y análisis de enriquecimiento funcional. Este conjunto de datos contiene las siguientes cinco categorías de datos:
| nombre | describir | Tamaño del conjunto de datos |
|---|---|---|
| EC$eset | Expresión genética normalizada en células endoteliales del cerebro y el corazón (3 réplicas) | 20644 x 7 |
| Lista de genes EC$ | Genes expresados diferencialmente (valor p ajustado <0,05) | 2039 x 7 |
| David EC$ | Resultados del análisis de enriquecimiento funcional de genes diferenciales utilizando DAVID. | 174 x 5 |
| Gen EC$ | Genes y logFC | 37 x 2 |
| Proceso EC$ | Vectores de características seleccionados para procesos biológicos enriquecidos. | 7 |
Queremos ver las vías enriquecidas con GO de genes expresados diferencialmente, pero antes de comenzar a dibujar debemos proporcionar datos que cumplan con los requisitos de formato.En términos generales, los datos necesarios para dibujar el gráfico los proporciona usted mismo, peroHay una función en este paquete.circle_datPuede ayudarnos a lidiar con el formato de datos.。circle_datPuede combinar los resultados del análisis de enriquecimiento funcional de genes seleccionados y sus valores logFC, principalmente para genes expresados diferencialmente.circle_dat El uso es muy sencillo, basta con leer dos datos. Los primeros datos contienen los resultados del análisis de enriquecimiento funcional, con al menos cuatro columnas (categoría de análisis de enriquecimiento funcional, vía, gen, valor p ajustado).El segundo dato es del gen seleccionado y su logFC, estos datos pueden ser la fuentelimmaLos resultados del análisis estadístico (Nota de biografías: asegúrese de prestar atención a dos archivosCómo se nombran los genesSea consistente, como todosGene symbol ). Veamos los formatos de datos mencionados anteriormente con ejemplos.
- #安装已发布的稳定版本
- #install.packages('GOplot')
- #安装github上的开发版本
- #install_github('wencke/wencke.github.io')
- #载入包
- library(GOplot)
- #读入包内自带的数据
- data(EC)
- #查看功能富集分析结果的数据格式
- head(EC$david)
-
- ## Category ID Term
- ## 1 BP GO:0007507 heart development
- ## 2 BP GO:0001944 vasculature development
- ## 3 BP GO:0001568 blood vessel development
- ## 4 BP GO:0048729 tissue morphogenesis
- ## 5 BP GO:0048514 blood vessel morphogenesis
- ## 6 BP GO:0051336 regulation of hydrolase activity
- ## Genes
- ## 1 DLC1, NRP2, NRP1, EDN1, PDLIM3, GJA1, TTN, GJA5, ZIC3, TGFB2, CERKL, GATA6, COL4A3BP, GAB1, SEMA3C, MKL2, SLC22A5, MB, PTPRJ, RXRA, VANGL2, MYH6, TNNT2, HHEX, MURC, MIB1, FOXC2, FOXC1, ADAM19, MYL2, TCAP, EGLN1, SOX9, ITGB1, CHD7, HEXIM1, PKD2, NFATC4, PCSK5, ACTC1, TGFBR2, NF1, HSPG2, SMAD3, TBX1, TNNI3, CSRP3, FOXP1, KCNJ8, PLN, TSC2, ATP6V0A1, TGFBR3, HDAC9
- ## 2 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, EFNB2, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 3 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 4 DLC1, ENAH, NRP1, PGF, ZIC2, TGFB2, CD44, ILK, SEMA3C, RET, AR, RXRA, VANGL2, LEF1, TNNT2, HHEX, MIB1, NCOA3, FOXC2, FOXC1, TGFB1I1, WNT5A, COBL, BBS4, FGFR3, TNC, BMPR2, CTNND1, EGLN1, NR3C1, SOX9, TCF7L1, IGF1R, FOXQ1, MACF1, HOXA5, BCL2, PLXND1, CAR2, ACTC1, TBX4, SMAD3, FZD3, SHANK3, FZD6, HOXB4, FREM2, TSC2, ZIC5, TGFBR3, APAF1
- ## 5 GNA13, CAV1, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, CDH2, MEIS1, WT1, TGFB2, WARS, PTK2, CERKL, APOE, CXCR4, ANG, SEMA3C, PLCD1, NOS2, MKL2, PLXND1, FIGF, FGF2, PTPRJ, TGFBR2, TBX4, NF1, TBX1, TNNI3, PNPLA6, VASH1, THY1, NUS1, MEOX2, ECSCR, AMOT, HBEGF, FOXC2, FOXC1, HDAC7
- ## 6 CAV1, XIAP, AGFG1, ADORA2A, TNNC1, TBC1D9, LEPR, ABHD5, EDN1, ASAP2, ASAP3, SMAP1, TBC1D12, ANG, TBC1D14, MTCH1, TBC1D13, TBC1D4, TBC1D30, DHCR24, HIP1, VAV3, NOS1, NF1, MYH6, RICTOR, TBC1D22A, THY1, PLCE1, RNF7, NDEL1, CHML, IFT57, ACAP2, TSC2, ERN1, APAF1, ARAP3, ARAP2, ARAP1, HTR2A, F2R
- ## adj_pval
- ## 1 0.000002170
- ## 2 0.000010400
- ## 3 0.000007620
- ## 4 0.000119000
- ## 5 0.000720000
- ## 6 0.001171166
-
- #查看基因的数据格式
- head(EC$genelist)
-
- ## ID logFC AveExpr t P.Value adj.P.Val B
- ## 1 Slco1a4 6.645388 1.2168670 88.65515 1.32e-18 2.73e-14 29.02715
- ## 2 Slc19a3 6.281525 1.1600468 69.95094 2.41e-17 2.49e-13 27.62917
- ## 3 Ddc 4.483338 0.8365231 65.57836 5.31e-17 3.65e-13 27.18476
- ## 4 Slco1c1 6.469384 1.3558865 59.87613 1.62e-16 8.34e-13 26.51242
- ## 5 Sema3c 5.515630 2.3252117 58.53141 2.14e-16 8.81e-13 26.33626
- ## 6 Slc38a3 4.761755 0.9218670 54.11559 5.58e-16 1.76e-12 25.70308
Después de comprender los dos formatos de datos de entrada, puede utilizarcirlce_datfunción para generar datos de dibujo.
- # 生成画图所需的数据格式
- circ <- circle_dat(EC$david, EC$genelist)
-
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
circEl objeto tiene ocho columnas de datos, a saber
categoría: BP (Proceso Biológico), CC (Componente Celular) o MF (Función Molecular)
ID: GO id (columna opcional, si desea utilizar una herramienta de análisis funcional que no se base en GO id, no puede seleccionar la columna ID; la ID aquí también puede ser KEGG ID)
término: vía GO
recuento: número de genes en cada vía
gen: nombre del gen - logFC: valor logFC de cada gen
adj_pval: valor p ajustado, las vías con adj_pval<0,05 se consideran significativamente enriquecidas
zscore: zscore no se refiere a un método de normalización estadística, sino que es un valor fácilmente calculado para estimar si es más probable que un proceso biológico (/función molecular/componente celular) disminuya (valor negativo) o aumente (valor positivo).El método de cálculo es el número de genes regulados positivamente menos el número de genes regulados negativamente dividido por la raíz cuadrada del número de genes en cada vía.
Cuando miramos los datos por primera vez, queremos mostrar tantas rutas como sea posible en el gráfico y también queremos encontrar rutas valiosas, por lo que necesitamos algunos parámetros para evaluar la importancia. Los gráficos de barras se usan a menudo para describir datos de muestra, por lo que podemos usar la función GOBar para crear rápidamente un gráfico de barras atractivo.
Primero, se genera directamente un gráfico de barras simple. El eje horizontal es.GO Terms, De acuerdo a suszscoreOrdena las barras; el eje vertical es-log(adj p-value);El color representazscore, el azul indicaz-scorees un valor negativo, es más probable que disminuya la expresión genética en la vía correspondiente, indicado en rojoz-score es un valor positivo, es más probable que aumente la expresión genética en la vía correspondiente. Si lo desea, el orden se puede cambiar estableciendo el parámetro order.by.zscore en FALSE, en cuyo caso las barras se ordenan según su importancia.
- # 生成简单的条形图
- GOBar(subset(circ, category == 'BP'))
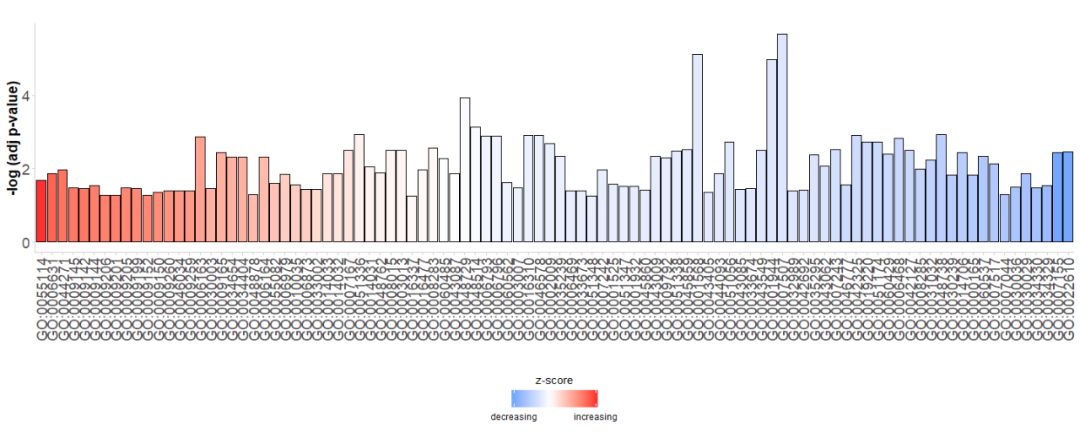
#GOBar(subset(circ, category == 'BP',order.by.zscore=FALSE))Además, cambie el parámetro de visualización para dibujar un gráfico de barras según la categoría del canal.
- #根据通路的类别来绘制条形图
- GOBar(circ, display = 'multiple')
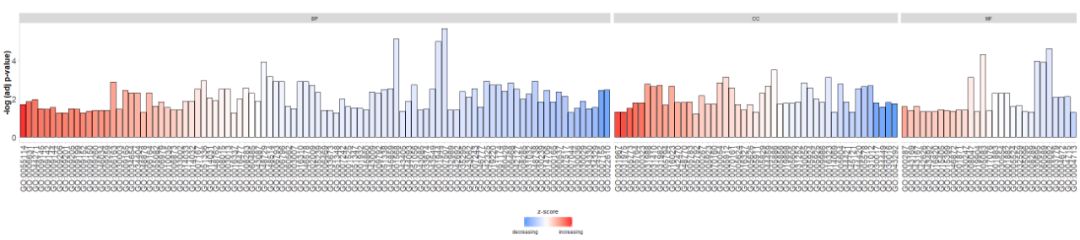
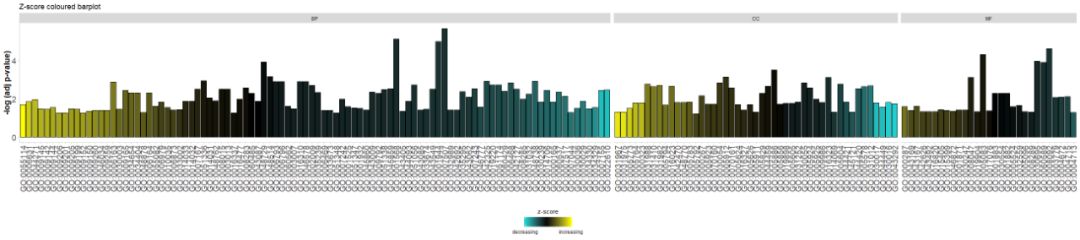
Agregar un título y usar parámetroszsc.colCambiarzscores color.
- # Facet the barplot, add a title and change the colour scale for the z-score
- GOBar(circ, display = 'multiple', title = 'Z-score coloured barplot', zsc.col = c('yellow', 'black', 'cyan'))
Los gráficos de barras son muy comunes y fáciles de entender, pero podemos utilizar gráficos de burbujas para mostrar más información sobre los datos.
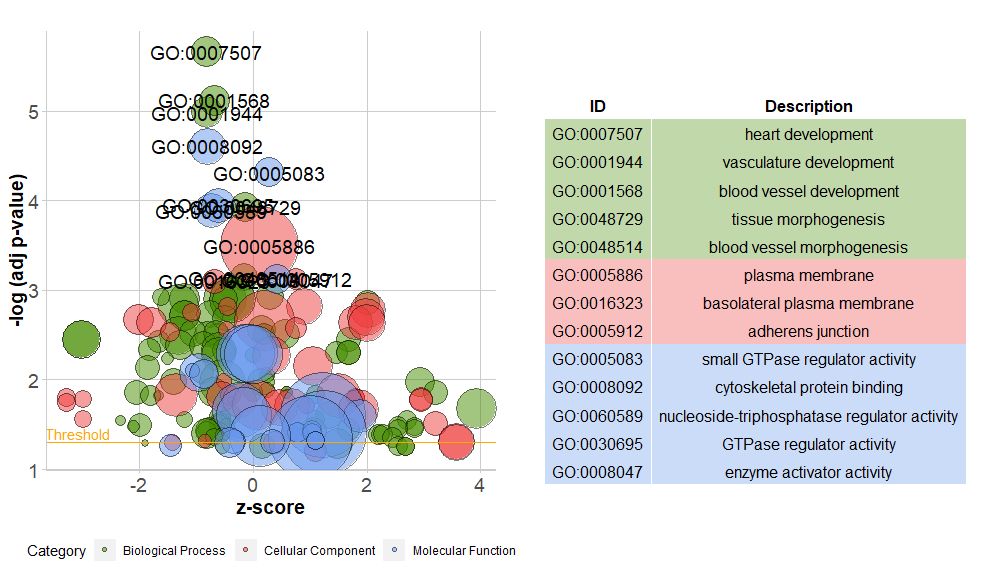
El eje horizontal eszscore;El eje vertical es-log(adj p-value), similar a un gráfico de barras, cuanto más alto es, más significativo es el enriquecimiento y el área del círculo está relacionada con la cantidad de genes en la vía correspondiente (circ$count ); el color corresponde a la categoría correspondiente a la vía, el verde es el proceso biológico, el rojo es el componente celular y el azul es la función molecular.Se puede ingresar por?GOBubble Consulte la página de ayuda de la función GOBubble para cambiar todos los parámetros de la imagen. De forma predeterminada, cada círculo está marcado con un ID de GO correspondiente y a la derecha también se muestra una tabla que muestra la relación correspondiente entre el ID de GO y el término GO.Los parámetros se pueden configurar mediantetable.legendparaFALSE para ocultarlo. Si desea mostrar la descripción de la ruta, establezca el ID del parámetro en FALSO.Sin embargo, debido al espacio limitado y a los círculos superpuestos, no todos los círculos están marcados, sólo los-log(adj p-value) > 3(el valor predeterminado es 5).
- # 生成泡泡图,并展示-log(adj p-value) > 3 的通路的GO ID
- GOBubble(circ, labels = 3)
Si desea agregar un título al gráfico de burbujas, o especificar el color del círculo y mostrar las rutas de cada categoría por separado y cambiar el umbral de ID de GO mostrado, puede agregar los siguientes parámetros:
GOBubble(circ, title = 'Bubble plot', colour = c('orange', 'darkred', 'gold'), display = 'multiple', labels = 3)
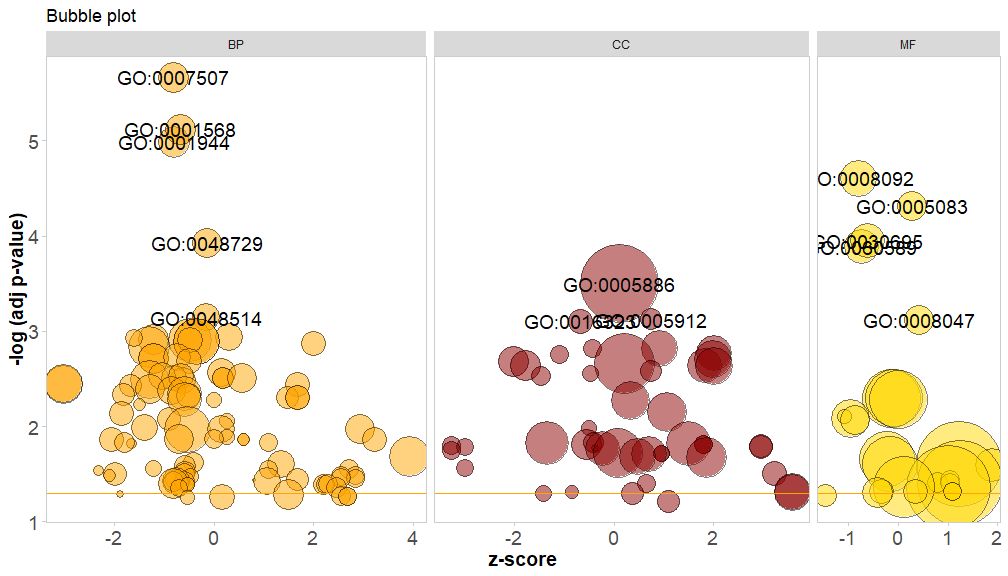
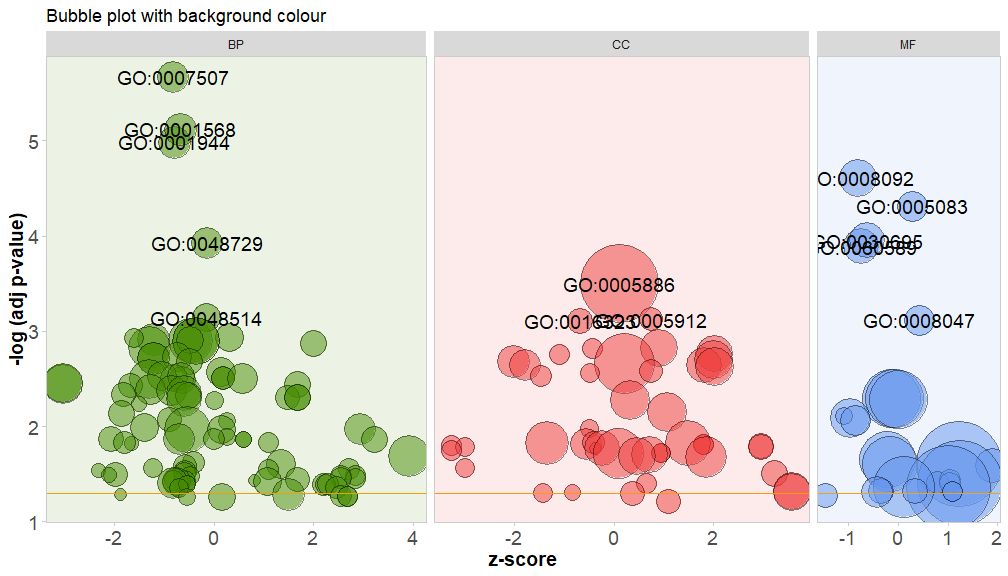
Colorea el fondo de la clase del canal configurando el parámetro bg.col en TRUE.
GOBubble(circ, title = 'Bubble plot with background colour', display = 'multiple', bg.col = T, labels = 3)
La nueva versión del paquete contiene una nueva función.reduce_overlap , esta función puede reducir el número de elementos redundantes, es decir, puede eliminar todas las vías cuya superposición de genes sea mayor o igual que el umbral establecido, y solo retener una vía de cada grupo como representante, independientemente de la visualización de todos. caminos en GO. Al reducir el número de términos redundantes, se mejora significativamente la legibilidad de los gráficos (como los gráficos de burbujas).
- # reduce_overlap,参数设置为0.75
- reduced_circ <- reduce_overlap(circ, overlap = 0.75)
-
- GOBubble(reduced_circ, labels = 2.8)
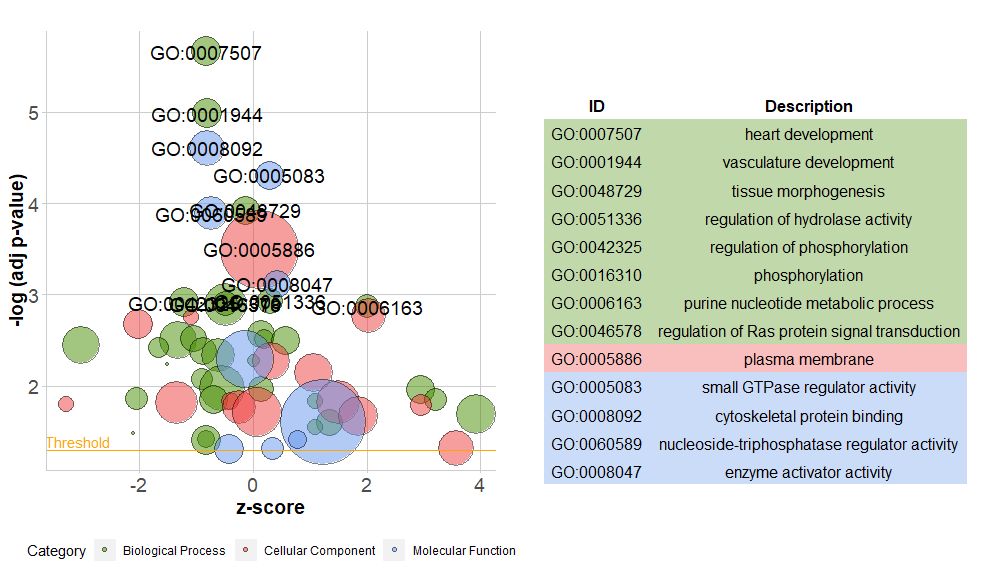
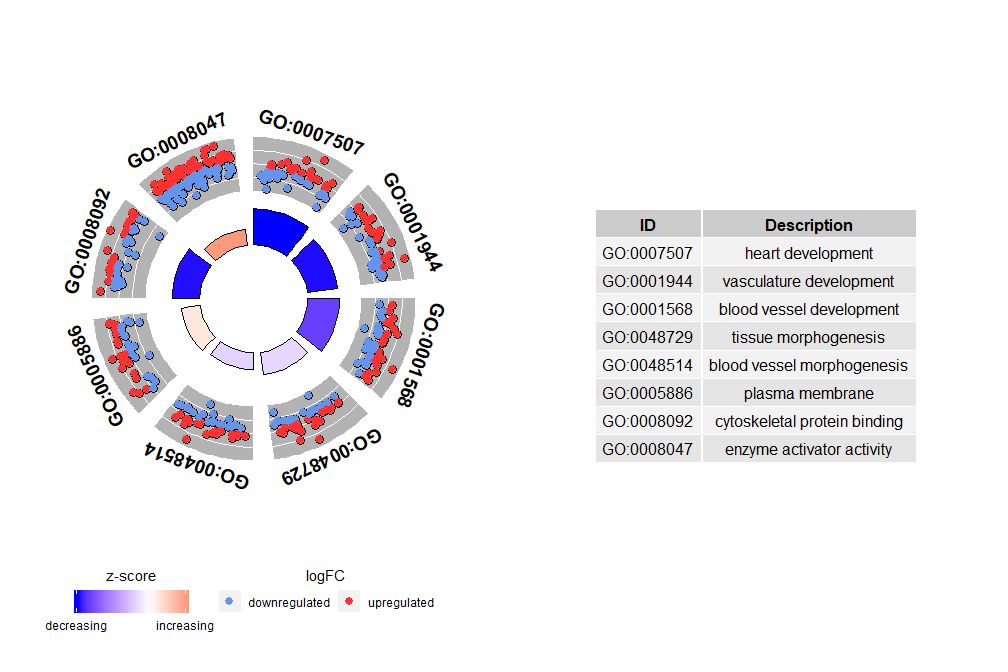
Aunque un gráfico que muestre toda la información puede ayudarnos a descubrir qué vías son más significativas, la realidad sigue dependiendo de las hipótesis e ideas que quieras confirmar con los datos, y las vías más importantes pueden no ser necesariamente las que te interesan. Por lo tanto, al seleccionar manualmente un valioso conjunto de vías (EC$process ), necesitamos un diagrama que nos muestre información más detallada sobre este conjunto específico de vías.Pero surge un problema al presentar estas cifras: a veces es difícil interpretarzscore Información proporcionada.Después de todo, este método de cálculo no es universal, como se muestra arriba, es simplemente el número de genes regulados positivamente menos el número de genes regulados negativamente dividido por la raíz cuadrada del número de genes en cada vía, usando.GOCircleEl gráfico resultante también enfatiza este hecho.
El círculo exterior del diagrama circular muestra el valor logFC de los genes de cada vía como puntos dispersos. Los círculos rojos indican regulación positiva y los azules indican regulación negativa.Se pueden utilizar parámetroslfc.col Cambiar el color. Esto también explica por qué en algunos casos rutas muy importantes tienen puntuaciones z cercanas a cero. Una puntuación z de cero no significa que el canal no sea importante. Simplemente muestra que zscore es una medida aproximada, porque obviamente zscore tampoco tiene en cuenta el nivel funcional y la dependencia de activación de genes individuales en los procesos biológicos.
GOCircle(circ)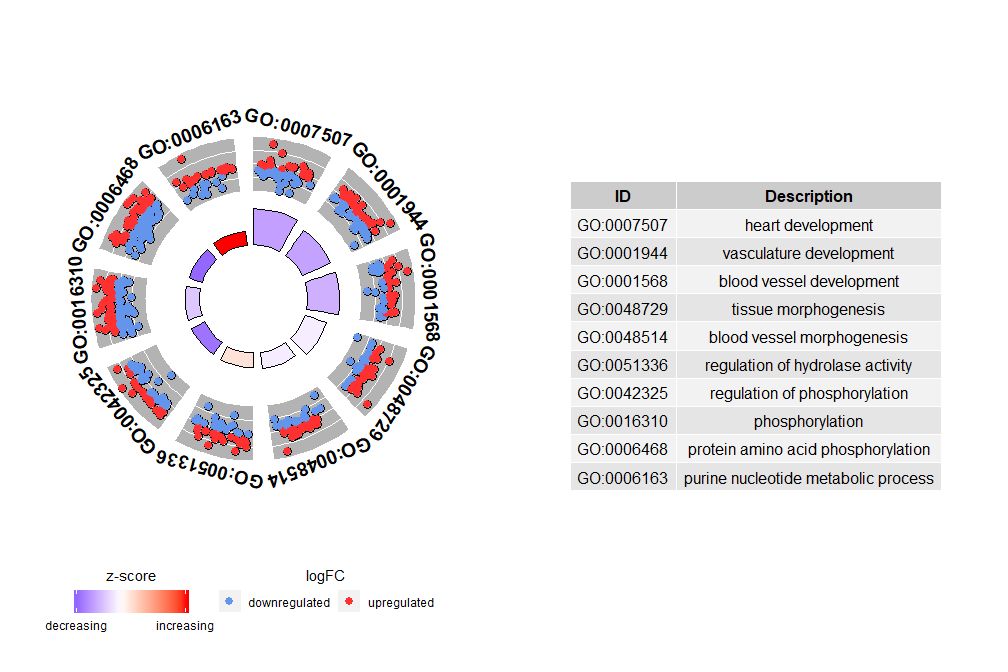
nsub Los parámetros pueden ser números establecidos o vectores de caracteres. Si es un vector de caracteres, contiene el ID de GO o la ruta a mostrar;
- # 生成特定通路的圈图
- IDs <- c('GO:0007507', 'GO:0001568', 'GO:0001944', 'GO:0048729', 'GO:0048514', 'GO:0005886', 'GO:0008092', 'GO:0008047')
- GOCircle(circ, nsub = IDs)
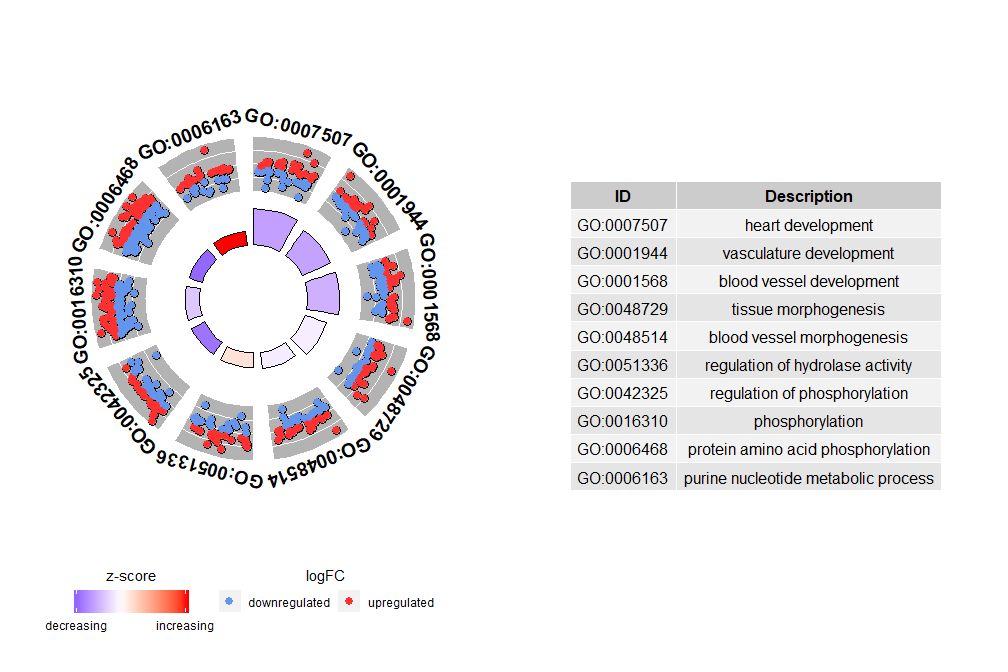
Si nsub es un vector numérico, el número define el número que se mostrará. Comienza desde la primera fila del marco de datos de entrada. Esta visualización solo funciona con datos más pequeños. El número máximo de canales por defecto es 12. Aunque se reduce el número de canales, aumenta la cantidad de información mostrada.
- # 圈图展示数据前十个通路
- GOCircle(circ, nsub = 10)
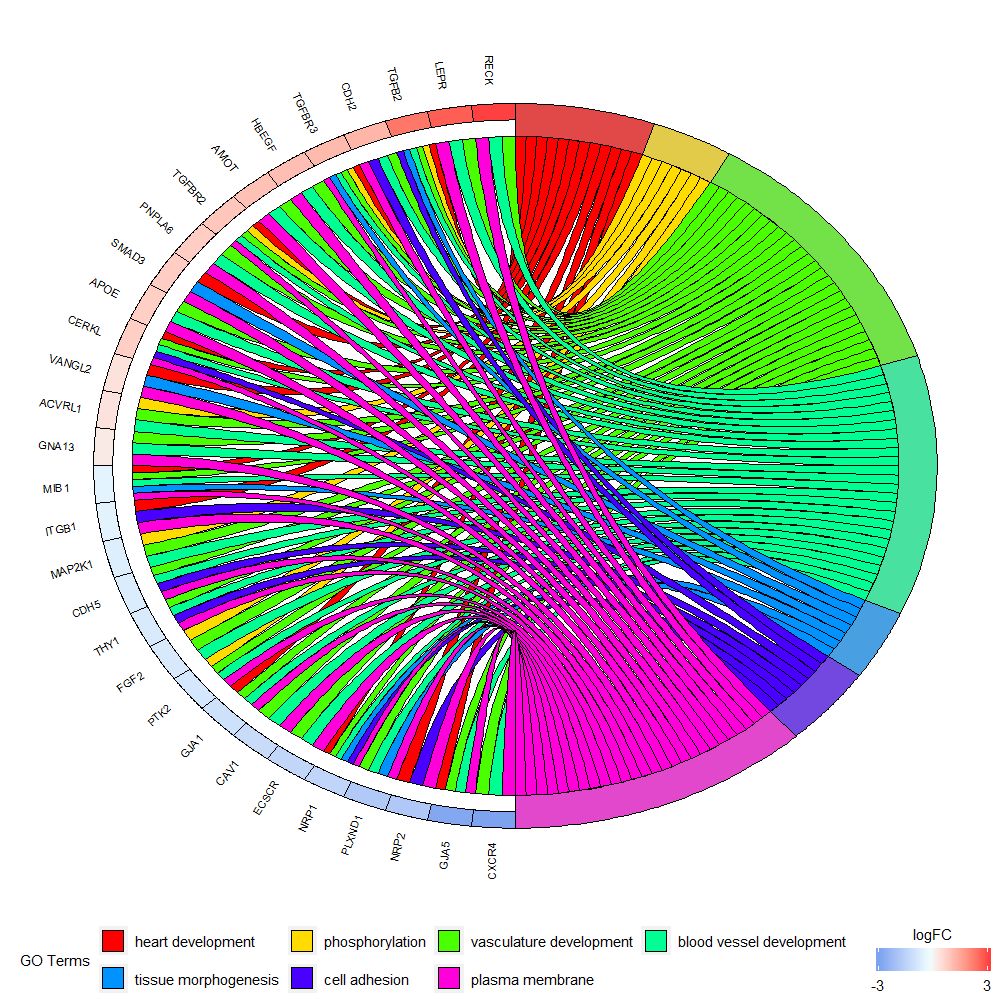
GOChord puede mostrar la relación entre genes y vías seleccionados y el logFC de los genes.Primero necesitas ingresar una matriz, que puedes construir tú mismo.0-1Matrix, también puedes usar funciones.chord_dat Construir. Esta función tiene tres parámetros: datos, genes y proceso, de los cuales los dos últimos parámetros deben tener al menos un parámetro.Entonces la funcióncircle_datCombine datos de expresión con resultados de análisis funcionales.
Los gráficos de barras y de burbujas pueden brindarle una primera impresión de los datos. Ahora, puede seleccionar algunos genes y vías que creemos que son valiosos. Aunque GOCircle agrega una capa para mostrar el valor de expresión de los genes en las vías, carece de información individual sobre. Relaciones entre genes y múltiples vías. No es fácil determinar si ciertos genes están relacionados con múltiples procesos. GOChord compensa las deficiencias de GOCircle. Las filas de los datos generados son genes y las columnas son vías. "0" significa que el gen no está asignado a la vía y "1" es lo contrario.
- # 找到感兴趣的的基因,这里我们以EC$genes为例
- head(EC$genes)
-
- ## ID logFC
- ## 1 PTK2 -0.6527904
- ## 2 GNA13 0.3711599
- ## 3 LEPR 2.6539788
- ## 4 APOE 0.8698346
- ## 5 CXCR4 -2.5647537
- ## 6 RECK 3.6926860
-
- # 获得感兴趣基因的通路
- EC$process
-
- ## [1] "heart development" "phosphorylation"
- ## [3] "vasculature development" "blood vessel development"
- ## [5] "tissue morphogenesis" "cell adhesion"
- ## [7] "plasma membrane"
-
- # 使用chord_dat构建矩阵
- chord <- chord_dat(circ, EC$genes, EC$process)
- head(chord)
-
- ## heart development phosphorylation vasculature development
- ## PTK2 0 1 1
- ## GNA13 0 0 1
- ## LEPR 0 0 1
- ## APOE 0 0 1
- ## CXCR4 0 0 1
- ## RECK 0 0 1
- ## blood vessel development tissue morphogenesis cell adhesion
- ## PTK2 1 0 0
- ## GNA13 1 0 0
- ## LEPR 1 0 0
- ## APOE 1 0 0
- ## CXCR4 1 0 0
- ## RECK 1 0 0
- ## plasma membrane logFC
- ## PTK2 1 -0.6527904
- ## GNA13 1 0.3711599
- ## LEPR 1 2.6539788
- ## APOE 1 0.8698346
- ## CXCR4 1 -2.5647537
- ## RECK 1 3.6926860
En el ejemplo, pasamos dos parámetros. Si solo se especifica el parámetro genes, el resultado es una lista de genes seleccionados y todas las construcciones de procesos con al menos un gen especificado.0-1matriz; si sólo se especificaprocessparámetros, el resultado es que todos los genes generan0-1 Matriz de genes asignados a al menos un proceso de la lista. Tenga en cuenta que especificar solo los genes y los parámetros del proceso puede generar una matriz 0-1 muy grande, lo que generará resultados de visualización confusos.
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
-
- # Generate the matrix with a list of selected genes
- chord_genes <- chord_dat(data = circ, genes = EC$genes)
- head(chord_genes)
-
- ## heart development vasculature development blood vessel development
- ## PTK2 0 1 1
- ## GNA13 0 1 1
- ## LEPR 0 1 1
- ## APOE 0 1 1
- ## CXCR4 0 1 1
Este gráfico está destinado a mostrar un subconjunto más pequeño de datos de alta dimensión. Hay principalmente dos parámetros que se pueden ajustar:gene.orderynlfc . El parámetro de genes se puede especificar como "logFC", "alfabético", "ninguno". De hecho, generalmente especificamos el parámetro de genes como logFC; el parámetro nlfc es uno de los parámetros más importantes de esta función, porque puede manejar cómo cada gen tiene 0 o más valores de logFC presentados en la matriz. Por lo tanto debemos especificar parámetros para evitar errores.
Por ejemplo, si tiene una matriz sin valores logFC, debe configurarnlfc=0 O realizar un análisis de expresión diferencial en genes en múltiples condiciones o lotes. En este caso, cada gen contiene múltiples valores de logFC y es necesario establecer el número de columna nlfc=logFC. El valor predeterminado es "1" porque se cree que la mayoría de las veces solo habrá un valor logFC por gen. Utilice el parámetro de espacio para definir el espacio entre rectángulos coloreados que representan logFC. El parámetro gene.size especifica el tamaño de fuente del nombre del gen y gene.space especifica el tamaño del espacio entre los nombres de los genes.
- chord <- chord_dat(data = circ, genes = EC$genes, process = EC$process)
- GOChord(chord, space = 0.02, gene.order = 'logFC', gene.space = 0.25, gene.size = 5)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
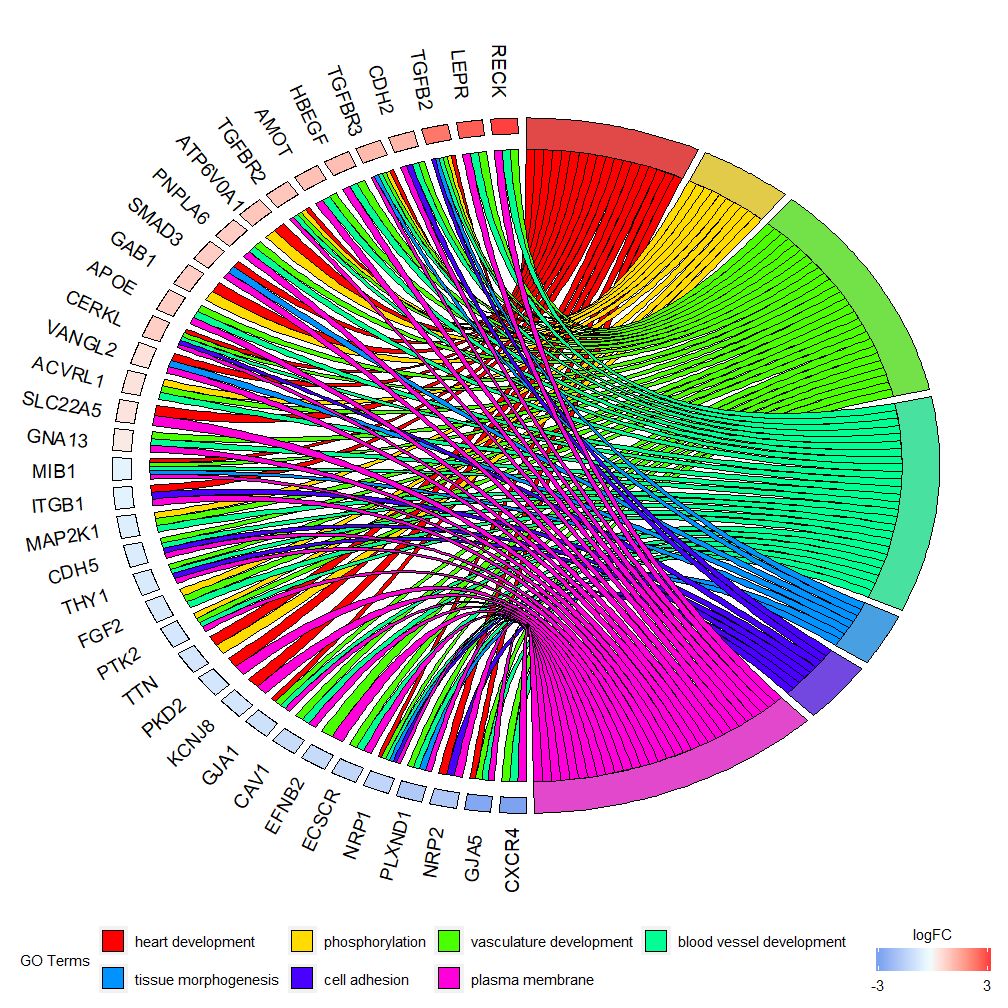
Se puede configurar según el valor logFCgene.order=‘logFC’ , clasifica los genes según sus valores logFC. A veces, la imagen puede llenarse un poco y esto se puede automatizar utilizando el parámetro de límite para reducir la cantidad de genes o vías mostradas. El límite es un vector con dos valores de corte (el valor predeterminado es c (0,0)). El primer valor especifica el número mínimo de vías a las que se debe asignar el gen. El segundo valor determina la cantidad de genes asignados a la vía.
- # 仅显示分配给至少三个通路的基因
- GOChord(chord, limit = c(3, 0), gene.order = 'logFC')
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
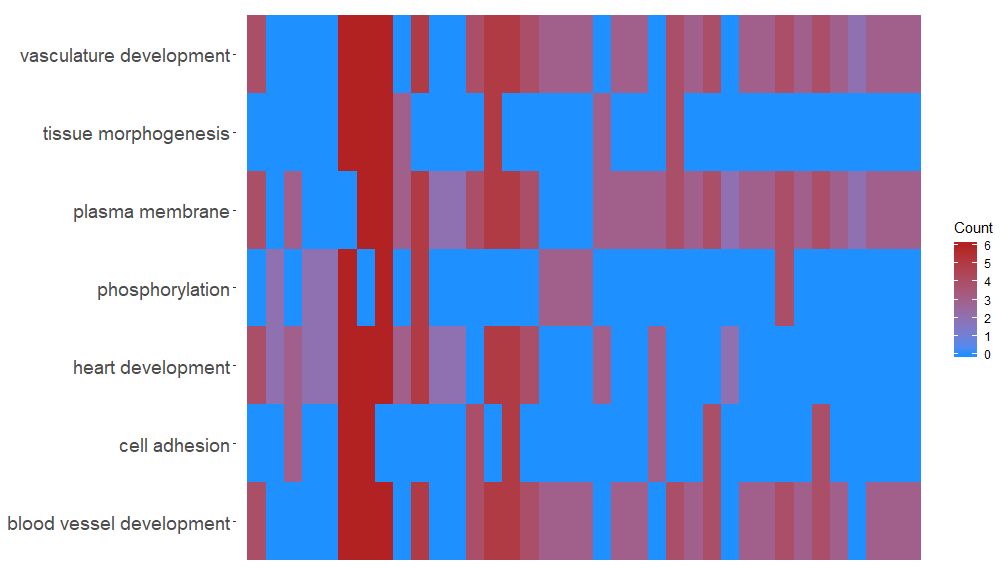
La función GOHeat puede mostrar la relación entre genes y vías mediante un mapa de calor, similar a GOChord. Los procesos biológicos se muestran horizontalmente y los genes se muestran verticalmente. Cada columna está dividida en pequeños rectángulos y el color generalmente depende del valor logFC. Además, se agruparon genes enriquecidos en vías funcionales similares. Hay dos modos para la selección del color del mapa de calor, según los parámetros de nlfc. Si nlfc = 0, el color es el número de vías enriquecidas para cada gen. Vea ejemplos para más detalles:
- # First, we use the chord object without logFC column to create the heatmap
- GOHeat(chord[,-8], nlfc = 0)
GOHeat(chord[,-8])El color corresponde al logFC del gen en caso de que nlfc = 1
GOHeat(chord, nlfc = 1, fill.col = c('red', 'yellow', 'green'))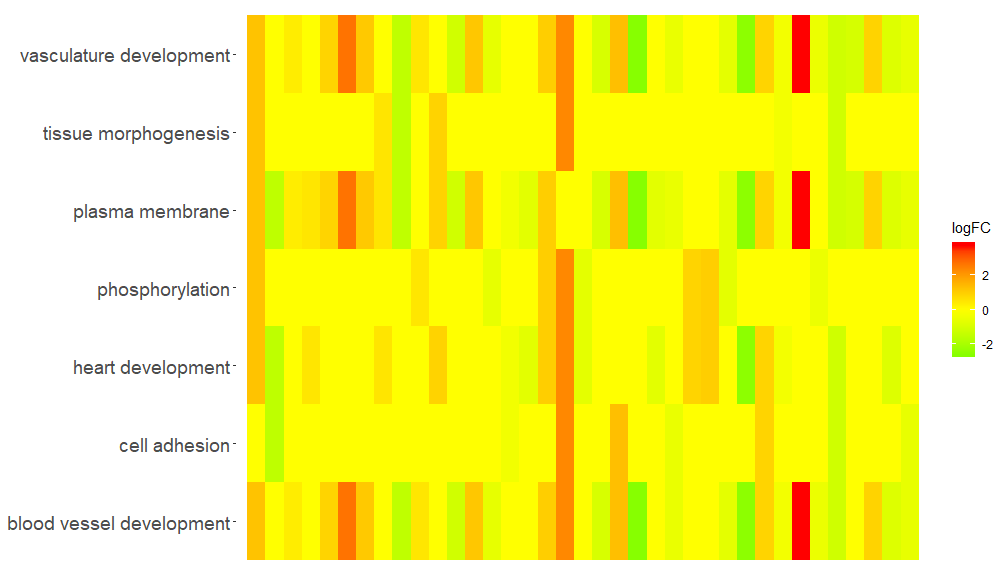
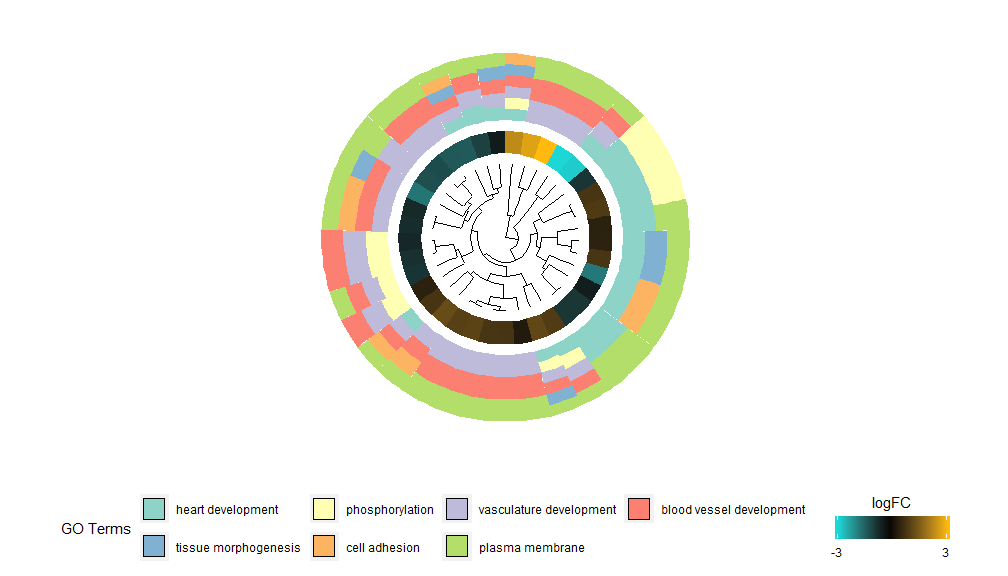
La idea detrás de la funcionalidad GOCluster es mostrar tanta información como sea posible. Aquí hay un ejemplo:
- GOCluster(circ, EC$process, clust.by = 'logFC', term.width = 2)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
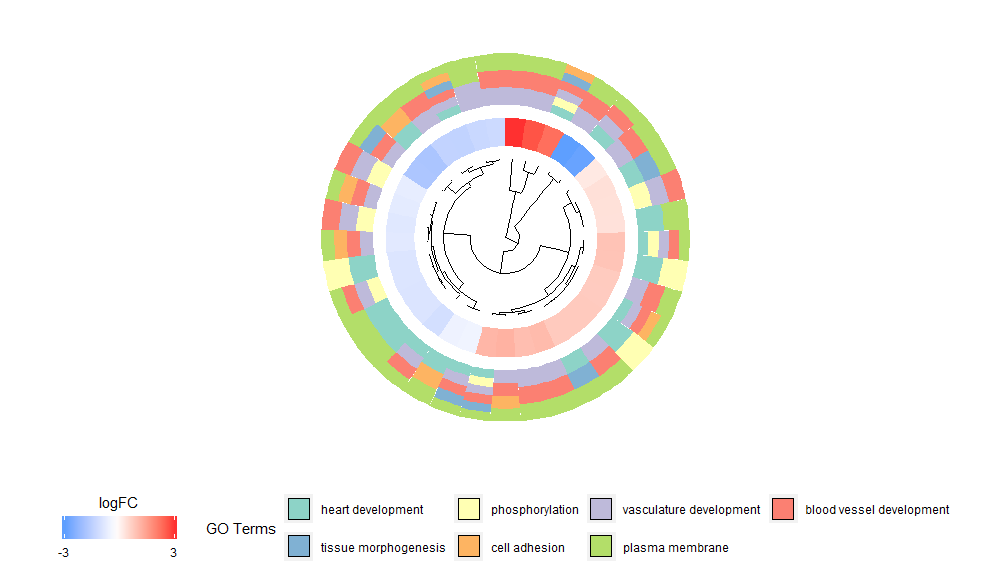
La agrupación jerárquica es un método popular de análisis de agrupación no supervisado para la expresión genética que garantiza una agrupación imparcial de genes por patrón de expresión, por lo que los grupos que se agrupan pueden contener múltiples grupos de genes corregulados o funcionalmente relacionados. GOCluster utiliza elhclust El método realiza una agrupación jerárquica de perfiles de expresión génica. Si desea cambiar la métrica de distancia o el algoritmo de agrupación, utilice los parámetros metric y clust respectivamente. El dendrograma resultante se puede convertir con la ayuda de ggdendro y visualizar con ggplot2. Elija un diseño circular, ya que no sólo es efectivo sino también visualmente atractivo. El primer círculo al lado del dendrograma representa el logFC del gen, que en realidad es la hoja del árbol de agrupación. Si está interesado en múltiples contrastes, puede modificar el parámetro nlfc; de forma predeterminada está establecido en "1", por lo que solo se dibuja un anillo. Los valores de logFC están codificados por colores utilizando una escala de colores definible por el usuario (lfc.col); el siguiente círculo representa la ruta asignada al gen; Para lucir bien, se ha reducido la cantidad de canales y el color de los canales se puede cambiar usando el parámetro term.col.aun disponible?GOCluster para ver cómo cambiar los parámetros. El parámetro más importante de esta función es cluster.by, que se puede especificar para agrupar por patrones de expresión genética ("logFC", como se muestra arriba) o categorías funcionales ("términos").
- GOCluster(circ, EC$process, clust.by = 'term', lfc.col = c('darkgoldenrod1', 'black', 'cyan1'))
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
Los diagramas de Venn se pueden utilizar para detectar relaciones entre varias listas de genes expresados diferencialmente o para explorar la intersección de múltiples genes de vías en análisis funcionales. Los diagramas de Venn no sólo muestran el número de genes superpuestos, sino también información sobre el patrón de expresión del gen (normalmente regulado positivamente, a menudo regulado negativamente o contrarregulado). Actualmente, se utilizan hasta tres conjuntos de datos como entrada. Los datos de entrada contienen al menos dos columnas: una para los nombres de los genes y otra para los valores logFC.
- l1 <- subset(circ, term == 'heart development', c(genes,logFC))
- l2 <- subset(circ, term == 'plasma membrane', c(genes,logFC))
- l3 <- subset(circ, term == 'tissue morphogenesis', c(genes,logFC))
- GOVenn(l1,l2,l3, label = c('heart development', 'plasma membrane', 'tissue morphogenesis'))
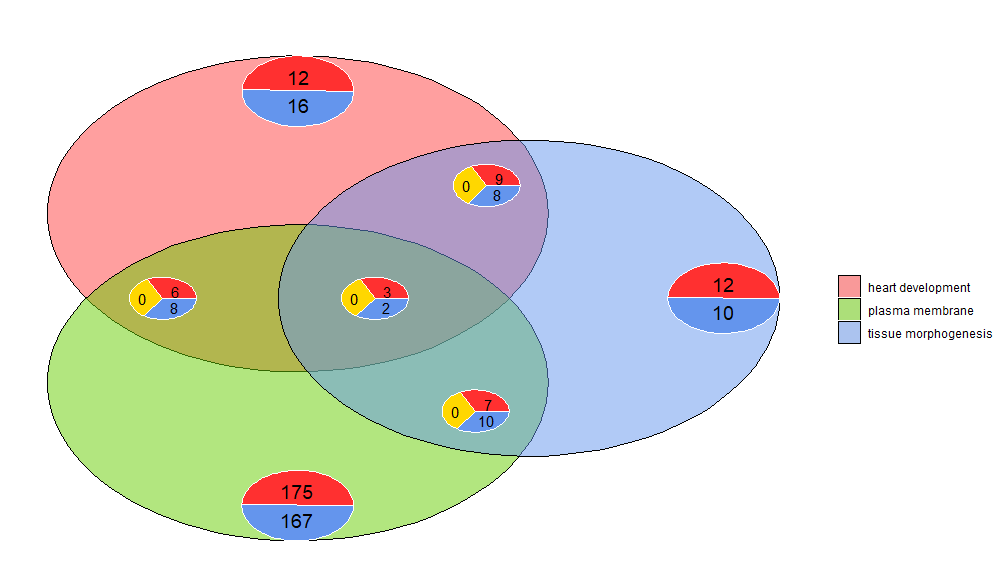
Por ejemplo, el desarrollo cardíaco y la morfogénesis tisular tienen 22 genes, 12 están regulados positivamente y 10 están regulados negativamente. Una cosa importante a tener en cuenta es que los gráficos circulares no muestran información redundante. Por lo tanto, si se comparan tres conjuntos de datos, los genes comunes a todos los conjuntos de datos (el gráfico circular del medio) no se incluyen en los otros gráficos circulares. Esta herramienta está disponible en shinyapp https://wwalter.shinyapps.io/Venn/, la herramienta web es más interactiva, el círculo tiene un área proporcional a la cantidad de genes en el conjunto de datos y el control deslizante se puede usar para mover el gráfico circular pequeño y cuenta con GOVenn con todas las opciones para cambiar el diseño del gráfico y también para descargar imágenes y listas de genes.
Página de inicio del software: https://wencke.github.io/

Se ha dedicado a la investigación de tecnología durante más de 30 años y domina varios lenguajes como java, linux, javascript, php, css, etc. Ha realizado muchas contribuciones en el campo del código abierto. Estación de documentación para desarrolladores para compartir algunos problemas en el desarrollo de tecnología para referencia futura.

Correo[email protected]