
내 연락처 정보
우편메소피아@프로톤메일.com
2024-07-12
한어Русский языкEnglishFrançaisIndonesianSanskrit日本語DeutschPortuguêsΕλληνικάespañolItalianoSuomalainenLatina
GOPlot 패키지는 생물학적 데이터의 시각화에 사용됩니다. 오히려 이 패키지는 기능 분석 결과와 함께 발현 데이터를 통합하고 시각화합니다.하지만 조심하세요이 패키지는 이러한 분석을 수행하는 데 사용할 수 없으며 결과를 시각화하는 데만 사용할 수 있습니다. . 과학의 모든 분야에서는 공간의 제약과 결과에 필요한 단순성으로 인해 사물을 현실적으로 기술하기가 어렵기 때문에 정보를 시각화하고 정보를 전달하기 위해 그림을 사용해야 합니다. 잘 디자인된 그래픽은 더 적은 공간에서 더 많은 정보를 제공합니다. 이 패키지의 아이디어는 사용자가 대량의 데이터를 빠르게 검토하고 데이터의 추세를 밝히고 데이터의 패턴과 상관 관계를 찾을 수 있도록 하는 것입니다.
데이터 시각화는 생물학적 질문에 대한 답을 찾고, 특정 가설을 판단하고, 다양한 문제를 조사하기 위한 다양한 각도를 발견하는 데 도움이 될 수 있습니다. 그리고 이 패키지의 플로팅 기능은 전체 데이터에서 시작하여 선택된 유전자의 하위 집합과 해당 경로로 끝나는 데이터의 계층 구조를 기반으로 개발되었습니다.
예를 들어 구체적으로 설명해보자.
우리는 GEO에서 제공되는 GOplot과 함께 제공되는 데이터를 호출합니다.GSE47067, 두 조직(뇌 및 심장)의 내피 세포에 대한 전사체 정보가 포함되어 있습니다. 자세한 내용은 Nolan et al. https://www.ncbi.nlm.nih.gov/pubmed/23871589를 참조하세요.데이터는 정규화되고 차별적으로 발현되는 유전자가 발견됩니다.을 누른 다음 DAVID 기능 주석 도구를 사용하세요(DAVID 주석 데이터는 느리게 업데이트되므로 지금은 권장되지 않습니다. 사용하는 것이 좋습니다).최고의 온라인 GO 농축 분석 도구인 Go East그리고단 한 단계로 농축 분석을 수행할 수 있는 이 웹사이트는 공개되기 전에 CNS 등에서 350회 이상 인용되었습니다.농축 분석 수행,하나의 기사로 GSEA를 마스터하고 매우 상세한 튜토리얼) 차등적으로 발현된 유전자의 유전자 주석(adjusted p-value < 0.05 ) 및 기능 강화 분석. 이 데이터 세트에는 다음과 같은 5가지 데이터 범주가 포함되어 있습니다.
| 이름 | 설명하다 | 데이터 세트 크기 |
|---|---|---|
| EC$이셋 | 뇌 및 심장 내피 세포의 정규화된 유전자 발현(3회 반복) | 20644 x 7 |
| EC$제네리스트 | 차별적으로 발현되는 유전자(조정된 p-값 < 0.05) | 2039 x 7 |
| EC$데이비드 | DAVID를 이용한 차등유전자의 기능적 농축 분석 결과 | 174 x 5 |
| EC$유전자 | 유전자와 logFC | 37 x 2 |
| EC$프로세스 | 풍부한 생물학적 과정을 위해 선택된 특징 벡터 | 7 |
우리는 차별적으로 발현된 유전자의 GO 강화 경로를 보고 싶지만 그리기를 시작하기 전에 형식 요구 사항을 충족하는 데이터를 제공해야 합니다.일반적으로 그래프를 그리는 데 필요한 데이터는 직접 제공하지만,이 패키지에는 기능이 있습니다circle_dat데이터 형식을 처리하는 데 도움이 될 수 있습니다.。circle_dat주로 차별적으로 발현되는 유전자에 대해 선택된 유전자의 기능적 농축 분석 결과와 해당 logFC 값을 결합할 수 있습니다.circle_dat 사용법은 매우 간단합니다. 두 개의 데이터만 읽으면 됩니다. 첫 번째 데이터에는 기능 강화 분석 결과가 포함되어 있으며 4개 이상의 열(기능 강화 분석 범주, 경로, 유전자, 조정된 p-값)이 포함되어 있습니다.두 번째 데이터는 선택된 유전자와 해당 logFC에 대한 것이며, 이 데이터는 소스가 될 수 있습니다.limma통계 분석 결과(약력 참고: 두 파일에 주의하세요.유전자의 이름은 어떻게 지정되나요?일관성을 유지하십시오.Gene symbol ). 위에서 언급한 데이터 형식을 예제와 함께 살펴보겠습니다.
- #安装已发布的稳定版本
- #install.packages('GOplot')
- #安装github上的开发版本
- #install_github('wencke/wencke.github.io')
- #载入包
- library(GOplot)
- #读入包内自带的数据
- data(EC)
- #查看功能富集分析结果的数据格式
- head(EC$david)
-
- ## Category ID Term
- ## 1 BP GO:0007507 heart development
- ## 2 BP GO:0001944 vasculature development
- ## 3 BP GO:0001568 blood vessel development
- ## 4 BP GO:0048729 tissue morphogenesis
- ## 5 BP GO:0048514 blood vessel morphogenesis
- ## 6 BP GO:0051336 regulation of hydrolase activity
- ## Genes
- ## 1 DLC1, NRP2, NRP1, EDN1, PDLIM3, GJA1, TTN, GJA5, ZIC3, TGFB2, CERKL, GATA6, COL4A3BP, GAB1, SEMA3C, MKL2, SLC22A5, MB, PTPRJ, RXRA, VANGL2, MYH6, TNNT2, HHEX, MURC, MIB1, FOXC2, FOXC1, ADAM19, MYL2, TCAP, EGLN1, SOX9, ITGB1, CHD7, HEXIM1, PKD2, NFATC4, PCSK5, ACTC1, TGFBR2, NF1, HSPG2, SMAD3, TBX1, TNNI3, CSRP3, FOXP1, KCNJ8, PLN, TSC2, ATP6V0A1, TGFBR3, HDAC9
- ## 2 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, EFNB2, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 3 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
- ## 4 DLC1, ENAH, NRP1, PGF, ZIC2, TGFB2, CD44, ILK, SEMA3C, RET, AR, RXRA, VANGL2, LEF1, TNNT2, HHEX, MIB1, NCOA3, FOXC2, FOXC1, TGFB1I1, WNT5A, COBL, BBS4, FGFR3, TNC, BMPR2, CTNND1, EGLN1, NR3C1, SOX9, TCF7L1, IGF1R, FOXQ1, MACF1, HOXA5, BCL2, PLXND1, CAR2, ACTC1, TBX4, SMAD3, FZD3, SHANK3, FZD6, HOXB4, FREM2, TSC2, ZIC5, TGFBR3, APAF1
- ## 5 GNA13, CAV1, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, CDH2, MEIS1, WT1, TGFB2, WARS, PTK2, CERKL, APOE, CXCR4, ANG, SEMA3C, PLCD1, NOS2, MKL2, PLXND1, FIGF, FGF2, PTPRJ, TGFBR2, TBX4, NF1, TBX1, TNNI3, PNPLA6, VASH1, THY1, NUS1, MEOX2, ECSCR, AMOT, HBEGF, FOXC2, FOXC1, HDAC7
- ## 6 CAV1, XIAP, AGFG1, ADORA2A, TNNC1, TBC1D9, LEPR, ABHD5, EDN1, ASAP2, ASAP3, SMAP1, TBC1D12, ANG, TBC1D14, MTCH1, TBC1D13, TBC1D4, TBC1D30, DHCR24, HIP1, VAV3, NOS1, NF1, MYH6, RICTOR, TBC1D22A, THY1, PLCE1, RNF7, NDEL1, CHML, IFT57, ACAP2, TSC2, ERN1, APAF1, ARAP3, ARAP2, ARAP1, HTR2A, F2R
- ## adj_pval
- ## 1 0.000002170
- ## 2 0.000010400
- ## 3 0.000007620
- ## 4 0.000119000
- ## 5 0.000720000
- ## 6 0.001171166
-
- #查看基因的数据格式
- head(EC$genelist)
-
- ## ID logFC AveExpr t P.Value adj.P.Val B
- ## 1 Slco1a4 6.645388 1.2168670 88.65515 1.32e-18 2.73e-14 29.02715
- ## 2 Slc19a3 6.281525 1.1600468 69.95094 2.41e-17 2.49e-13 27.62917
- ## 3 Ddc 4.483338 0.8365231 65.57836 5.31e-17 3.65e-13 27.18476
- ## 4 Slco1c1 6.469384 1.3558865 59.87613 1.62e-16 8.34e-13 26.51242
- ## 5 Sema3c 5.515630 2.3252117 58.53141 2.14e-16 8.81e-13 26.33626
- ## 6 Slc38a3 4.761755 0.9218670 54.11559 5.58e-16 1.76e-12 25.70308
두 가지 입력 데이터 형식을 이해한 후 다음을 사용할 수 있습니다.cirlce_dat도면 데이터를 생성하는 기능입니다.
- # 生成画图所需的数据格式
- circ <- circle_dat(EC$david, EC$genelist)
-
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
circ객체에는 8개의 데이터 열이 있습니다.
카테고리: BP(생물학적 과정), CC(세포 구성 요소) 또는 MF(분자 기능)
ID: GO id(선택 열, GO id를 기반으로 하지 않는 기능 분석 도구를 사용하려는 경우 ID 열을 선택할 수 없습니다. 여기의 ID는 KEGG ID일 수도 있습니다)
용어: GO 경로
개수: 각 경로의 유전자 수
gene: 유전자 이름 - logFC: 각 유전자의 logFC 값
adj_pval: 조정된 p 값, adj_pval<0.05인 경로는 상당히 강화된 것으로 간주됩니다.
zscore: zscore는 통계적 정규화 방법을 의미하지 않으며, 생물학적 과정(/분자기능/세포성분)이 감소(음수 값) 또는 증가(양수 값)할 가능성이 높은지 추정하기 위해 쉽게 계산된 값입니다.계산 방법은 상향 조절된 유전자 수에서 하향 조절된 유전자 수를 뺀 값을 각 경로의 유전자 수의 제곱근으로 나눈 값입니다.
처음 데이터를 볼 때 그래프를 통해 최대한 많은 경로를 보여주고 싶고, 가치 있는 경로도 찾고 싶기 때문에 중요성을 평가하기 위한 몇 가지 매개변수가 필요합니다. 막대 차트는 샘플 데이터를 설명하는 데 자주 사용되므로 GOBar 기능을 사용하면 보기 좋은 막대 차트를 빠르게 만들 수 있습니다.
먼저 간단한 막대 차트가 직접 생성됩니다.GO Terms, 그들의 말에 따르면zscore막대를 정렬합니다.-log(adj p-value);색상은 다음을 나타냅니다.zscore, 파란색은z-score음수 값이면 해당 경로의 유전자 발현이 감소할 가능성이 더 높으며 빨간색으로 표시됩니다.z-score 양수 값일수록 해당 경로의 유전자 발현이 증가할 가능성이 더 높습니다. 원하는 경우 order.by.zscore 매개변수를 FALSE로 설정하여 순서를 변경할 수 있습니다. 이 경우 막대는 중요도에 따라 정렬됩니다.
- # 生成简单的条形图
- GOBar(subset(circ, category == 'BP'))
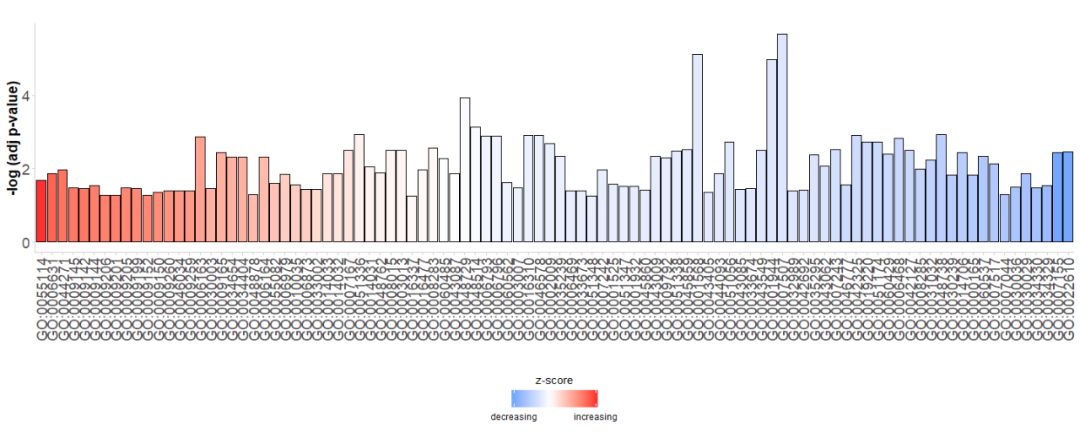
#GOBar(subset(circ, category == 'BP',order.by.zscore=FALSE))또한, 채널 카테고리에 따라 막대 차트를 그릴 수 있도록 표시 매개변수를 변경합니다.
- #根据通路的类别来绘制条形图
- GOBar(circ, display = 'multiple')
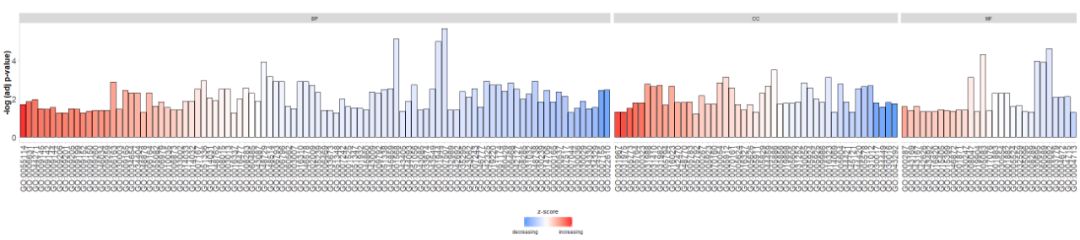
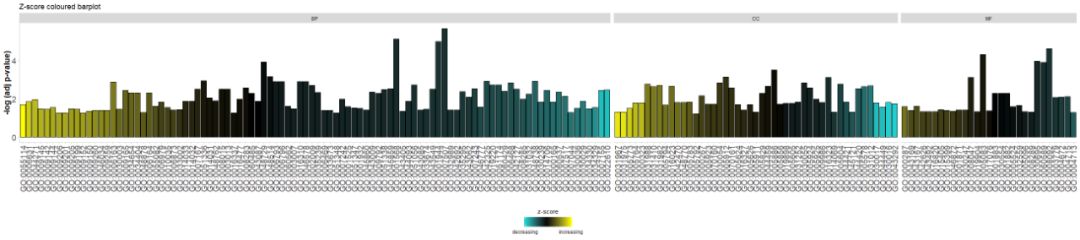
제목 추가 및 매개변수 사용zsc.col변화zscore색깔.
- # Facet the barplot, add a title and change the colour scale for the z-score
- GOBar(circ, display = 'multiple', title = 'Z-score coloured barplot', zsc.col = c('yellow', 'black', 'cyan'))
막대 차트는 매우 일반적이고 이해하기 쉽지만 버블 차트를 사용하여 데이터에 대한 더 많은 정보를 표시할 수 있습니다.
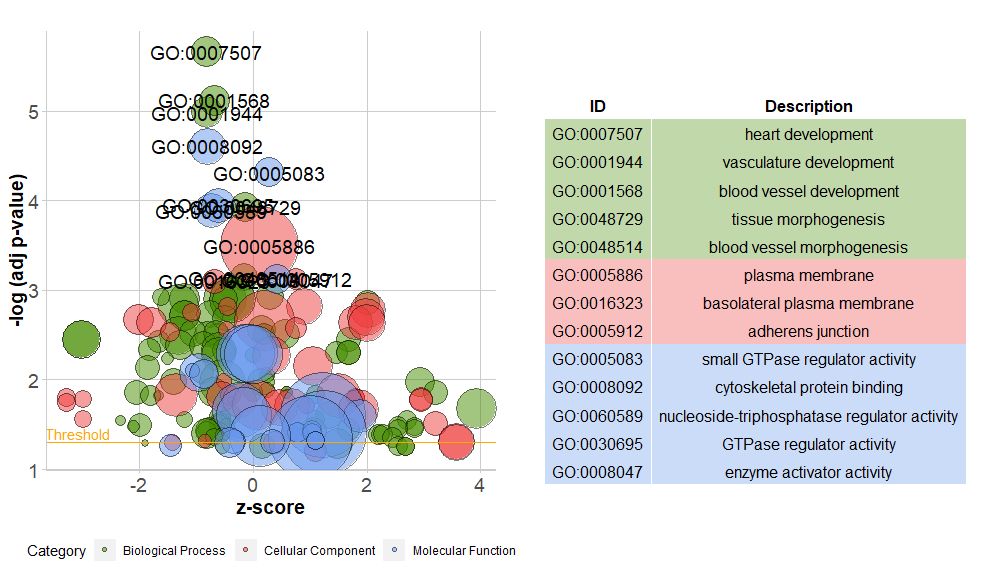
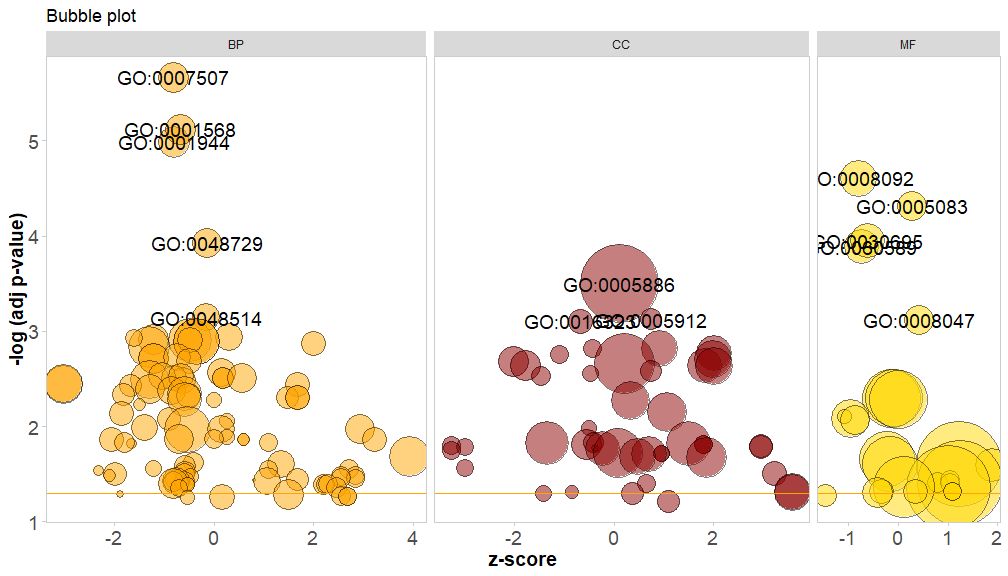
가로축은zscore;세로축은-log(adj p-value), 막대 차트와 유사하게 높을수록 농축도가 더 중요하며 해당 경로의 유전자 수와 관련이 있습니다.circ$count ) 색상은 경로에 해당하는 범주에 해당하고 녹색은 생물학적 과정, 빨간색은 세포 구성 요소, 파란색은 분자 기능입니다.로 입력 가능?GOBubble 이미지의 매개변수를 모두 변경하려면 GOBubble 기능의 도움말 페이지를 참조하세요. 기본적으로 각 원에는 해당 GO ID가 표시되어 있으며 GO ID와 GO 용어 간의 해당 관계를 보여주는 표도 오른쪽에 표시됩니다.매개변수는 다음에 의해 설정될 수 있습니다.table.legend~을 위한FALSE 그것을 숨기기 위해. 경로 설명을 표시하려면 매개변수 ID를 FALSE로 설정하세요.그러나 제한된 공간과 겹치는 원으로 인해 모든 원이 표시되지는 않습니다.-log(adj p-value) > 3(기본값은 5입니다).
- # 生成泡泡图,并展示-log(adj p-value) > 3 的通路的GO ID
- GOBubble(circ, labels = 3)
버블 차트에 제목을 추가하거나 원의 색상을 지정하고 각 범주의 경로를 별도로 표시하고 표시되는 GO ID 임계값을 변경하려는 경우 다음 매개변수를 추가할 수 있습니다.
GOBubble(circ, title = 'Bubble plot', colour = c('orange', 'darkred', 'gold'), display = 'multiple', labels = 3)
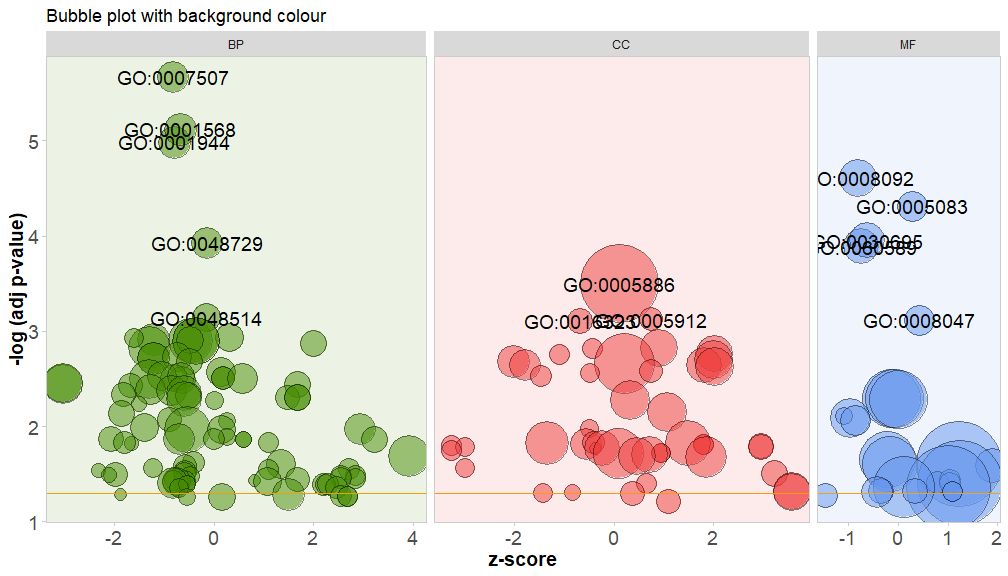
bg.col 매개변수를 TRUE로 설정하여 채널 클래스의 배경색을 지정합니다.
GOBubble(circ, title = 'Bubble plot with background colour', display = 'multiple', bg.col = T, labels = 3)
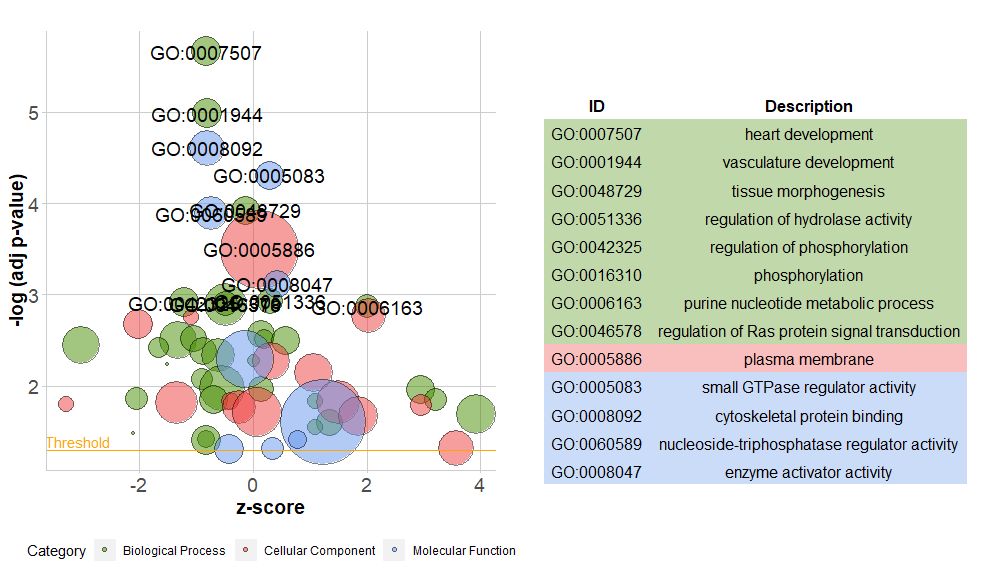
새 버전의 패키지에는 새로운 기능이 포함되어 있습니다.reduce_overlap , 이 기능은 중복 항목 수를 줄일 수 있습니다. 즉, 유전자 중복이 설정된 임계값보다 크거나 같은 모든 경로를 삭제할 수 있으며, 모든 표시에 관계없이 각 그룹에서 하나의 경로만 대표로 유지할 수 있습니다. GO의 경로. 중복되는 항의 수를 줄임으로써 도표(예: 버블 도표)의 가독성이 크게 향상됩니다.
- # reduce_overlap,参数设置为0.75
- reduced_circ <- reduce_overlap(circ, overlap = 0.75)
-
- GOBubble(reduced_circ, labels = 2.8)
모든 정보를 보여주는 그래프는 어떤 경로가 가장 의미 있는지 발견하는 데 도움이 될 수 있지만 현실은 여전히 데이터로 확인하려는 가설과 아이디어에 따라 달라지며 가장 중요한 경로가 반드시 관심 있는 경로가 아닐 수도 있습니다. 따라서 귀중한 경로 세트를 수동으로 선택하는 경우(EC$process ), 이 특정 경로 세트에 대한 더 자세한 정보를 표시하려면 다이어그램이 필요합니다.그러나 이러한 수치를 제시하면 문제가 발생합니다. 때로는 해석하기 어렵습니다.zscore 제공된 정보.결국, 이 계산 방법은 보편적이지 않습니다. 위에 표시된 것처럼 이는 단순히 상향 조절된 유전자 수에서 하향 조절된 유전자 수를 뺀 값을 각 경로의 유전자 수의 제곱근으로 나눈 값입니다.GOCircle결과 그래프도 이 사실을 강조합니다.
원 다이어그램의 바깥쪽 원은 각 경로의 유전자의 logFC 값을 산점도로 표시합니다. 빨간색 원은 상향 조절을 나타내고 파란색 원은 하향 조절을 나타냅니다.매개변수를 사용할 수 있습니다.lfc.col 색상을 변경합니다. 이는 또한 어떤 경우에 매우 중요한 경로의 zscore가 0에 가까운 이유를 설명합니다. zscore가 0이라고 해서 채널이 중요하지 않다는 의미는 아닙니다. 이는 zscore가 대략적인 측정값임을 보여줍니다. 왜냐하면 분명히 zscore는 생물학적 과정에서 개별 유전자의 기능적 수준과 활성화 의존성을 고려하지 않기 때문입니다.
GOCircle(circ)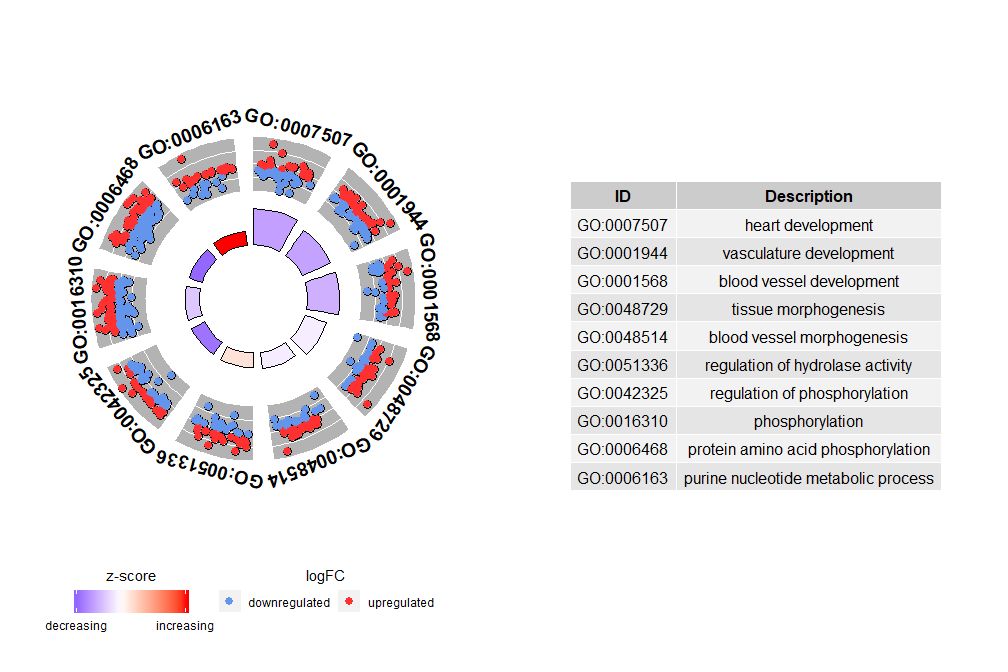
nsub 매개변수는 숫자 또는 문자형 벡터로 설정할 수 있습니다. 문자형 벡터인 경우 표시할 GO ID 또는 경로가 포함됩니다.
- # 生成特定通路的圈图
- IDs <- c('GO:0007507', 'GO:0001568', 'GO:0001944', 'GO:0048729', 'GO:0048514', 'GO:0005886', 'GO:0008092', 'GO:0008047')
- GOCircle(circ, nsub = IDs)
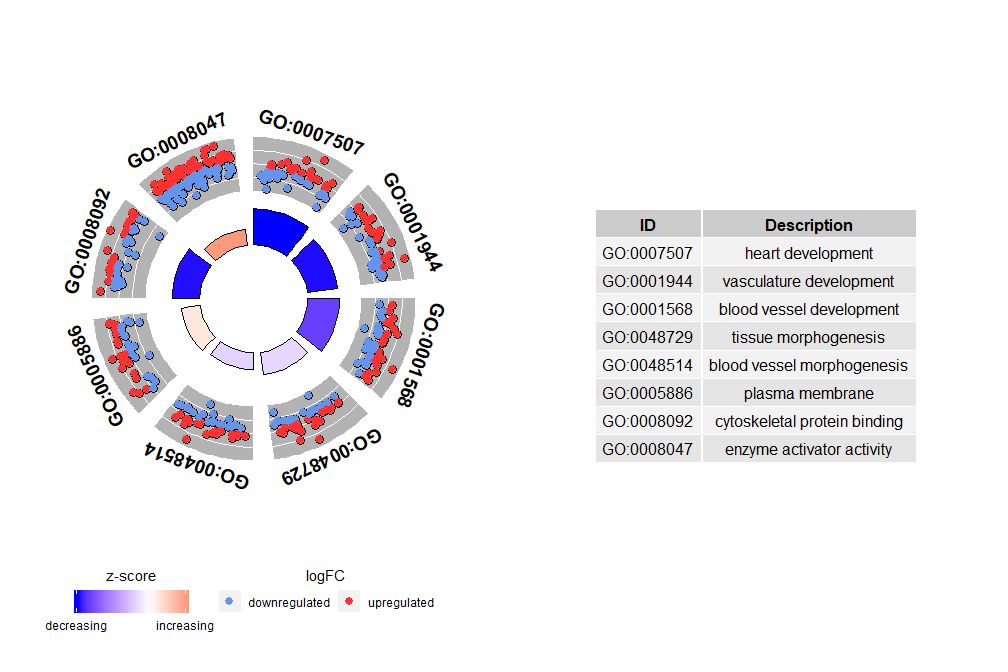
nsub가 숫자형 벡터인 경우 숫자는 표시할 숫자를 정의합니다. 입력 데이터 프레임의 첫 번째 행부터 시작됩니다. 이 시각화는 더 작은 데이터에서만 작동합니다. 최대 채널 수는 기본적으로 12개입니다. 채널 수는 줄어들지만 표시되는 정보의 양은 늘어납니다.
- # 圈图展示数据前十个通路
- GOCircle(circ, nsub = 10)
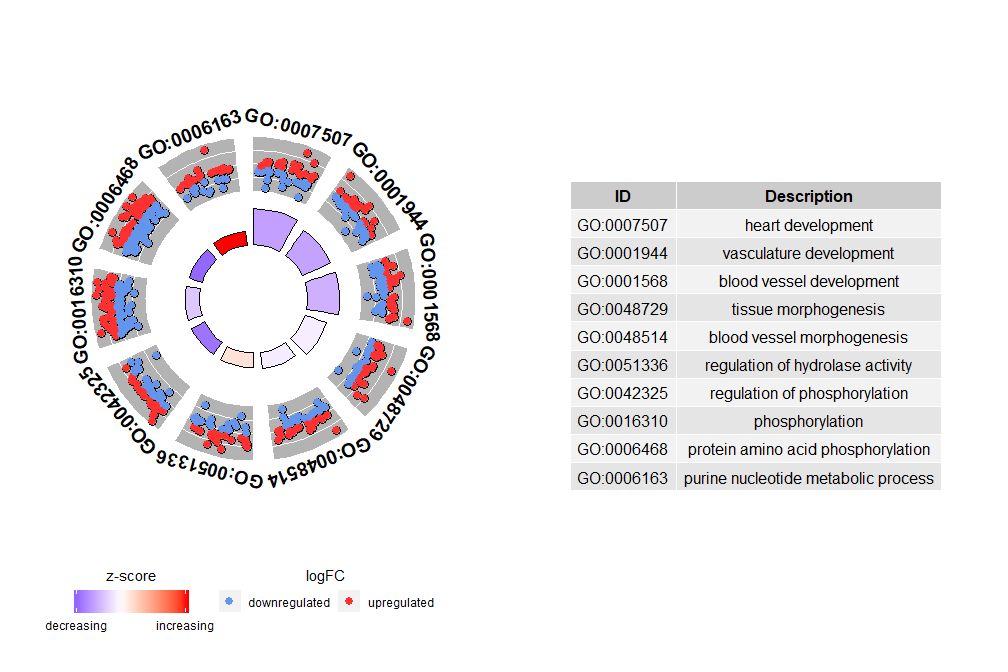
GOChord는 선택된 유전자와 경로, 그리고 유전자의 logFC 사이의 관계를 표시할 수 있습니다.먼저 직접 작성할 수 있는 행렬을 입력해야 합니다.0-1매트릭스, 함수를 사용할 수도 있습니다chord_dat 건설하다. 이 함수에는 데이터, 유전자, 프로세스의 세 가지 매개변수가 있으며, 그 중 마지막 두 매개변수에는 최소한 하나의 매개변수가 있어야 합니다.그런 다음 기능circle_dat발현 데이터를 기능 분석 결과와 결합합니다.
막대 차트와 버블 차트는 데이터에 대한 첫인상을 줄 수 있습니다. 이제 가치 있다고 생각되는 일부 유전자와 경로를 선택할 수 있습니다. GOCircle은 경로에 유전자의 발현 값을 표시하기 위해 레이어를 추가하지만 단일 정보가 부족합니다. 유전자와 다중 경로 사이의 관계에 대해. 특정 유전자가 여러 과정에 연결되어 있는지 알아내는 것은 쉽지 않습니다. GOChord는 GOCircle의 단점을 보완합니다. 생성된 데이터의 행은 유전자이고 열은 경로입니다. "0"은 유전자가 경로에 할당되지 않았음을 의미하고 "1"은 반대입니다.
- # 找到感兴趣的的基因,这里我们以EC$genes为例
- head(EC$genes)
-
- ## ID logFC
- ## 1 PTK2 -0.6527904
- ## 2 GNA13 0.3711599
- ## 3 LEPR 2.6539788
- ## 4 APOE 0.8698346
- ## 5 CXCR4 -2.5647537
- ## 6 RECK 3.6926860
-
- # 获得感兴趣基因的通路
- EC$process
-
- ## [1] "heart development" "phosphorylation"
- ## [3] "vasculature development" "blood vessel development"
- ## [5] "tissue morphogenesis" "cell adhesion"
- ## [7] "plasma membrane"
-
- # 使用chord_dat构建矩阵
- chord <- chord_dat(circ, EC$genes, EC$process)
- head(chord)
-
- ## heart development phosphorylation vasculature development
- ## PTK2 0 1 1
- ## GNA13 0 0 1
- ## LEPR 0 0 1
- ## APOE 0 0 1
- ## CXCR4 0 0 1
- ## RECK 0 0 1
- ## blood vessel development tissue morphogenesis cell adhesion
- ## PTK2 1 0 0
- ## GNA13 1 0 0
- ## LEPR 1 0 0
- ## APOE 1 0 0
- ## CXCR4 1 0 0
- ## RECK 1 0 0
- ## plasma membrane logFC
- ## PTK2 1 -0.6527904
- ## GNA13 1 0.3711599
- ## LEPR 1 2.6539788
- ## APOE 1 0.8698346
- ## CXCR4 1 -2.5647537
- ## RECK 1 3.6926860
예제에서는 두 개의 매개변수를 전달했습니다. gene 매개변수만 지정된 경우 결과는 선택된 유전자 목록과 최소한 하나의 지정된 유전자가 있는 모든 프로세스 구성입니다.0-1매트릭스만 지정된 경우process매개변수의 결과는 모든 유전자가 생성된다는 것입니다.0-1 목록에 있는 하나 이상의 프로세스에 할당된 유전자 매트릭스입니다. 유전자와 프로세스 매개변수만 지정하면 매우 큰 0-1 행렬이 생성되어 시각화 결과가 혼란스러울 수 있습니다.
- head(circ)
-
- ## category ID term count genes logFC adj_pval
- ## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
- ## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
- ## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
- ## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
- ## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
- ## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
- ## zscore
- ## 1 -0.8164966
- ## 2 -0.8164966
- ## 3 -0.8164966
- ## 4 -0.8164966
- ## 5 -0.8164966
- ## 6 -0.8164966
-
- # Generate the matrix with a list of selected genes
- chord_genes <- chord_dat(data = circ, genes = EC$genes)
- head(chord_genes)
-
- ## heart development vasculature development blood vessel development
- ## PTK2 0 1 1
- ## GNA13 0 1 1
- ## LEPR 0 1 1
- ## APOE 0 1 1
- ## CXCR4 0 1 1
이 차트는 고차원 데이터의 작은 하위 집합을 표시하기 위한 것입니다. 조정할 수 있는 매개변수는 주로 두 가지입니다.gene.order그리고nlfc . 유전자 매개변수는 'logFC', '알파벳순', '없음'으로 지정할 수 있습니다. 실제로 우리는 일반적으로 유전자 매개변수를 logFC로 지정합니다. nlfc 매개변수는 이 함수의 가장 중요한 매개변수 중 하나입니다. 이는 각 유전자가 행렬에 표시되는 0개 이상의 logFC 값을 처리할 수 있기 때문입니다. 따라서 오류를 방지하려면 매개변수를 지정해야 합니다.
예를 들어, logFC 값이 없는 행렬이 있는 경우 다음을 설정해야 합니다.nlfc=0 ; 또는 여러 조건 또는 배치에서 유전자에 대한 차등 발현 분석을 수행합니다. 이 경우 각 유전자에는 여러 개의 logFC 값이 포함되어 있으며 nlfc=logFC 열 번호를 설정해야 합니다. 대부분의 경우 유전자당 하나의 logFC 값만 있을 것으로 간주되기 때문에 기본값은 "1"입니다. logFC를 나타내는 색상이 지정된 직사각형 사이의 공간을 정의하려면 space 매개변수를 사용하십시오. gene.size 매개변수는 유전자 이름의 글꼴 크기를 지정하고, gene.space는 유전자 이름 사이의 공백 크기를 지정합니다.
- chord <- chord_dat(data = circ, genes = EC$genes, process = EC$process)
- GOChord(chord, space = 0.02, gene.order = 'logFC', gene.space = 0.25, gene.size = 5)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
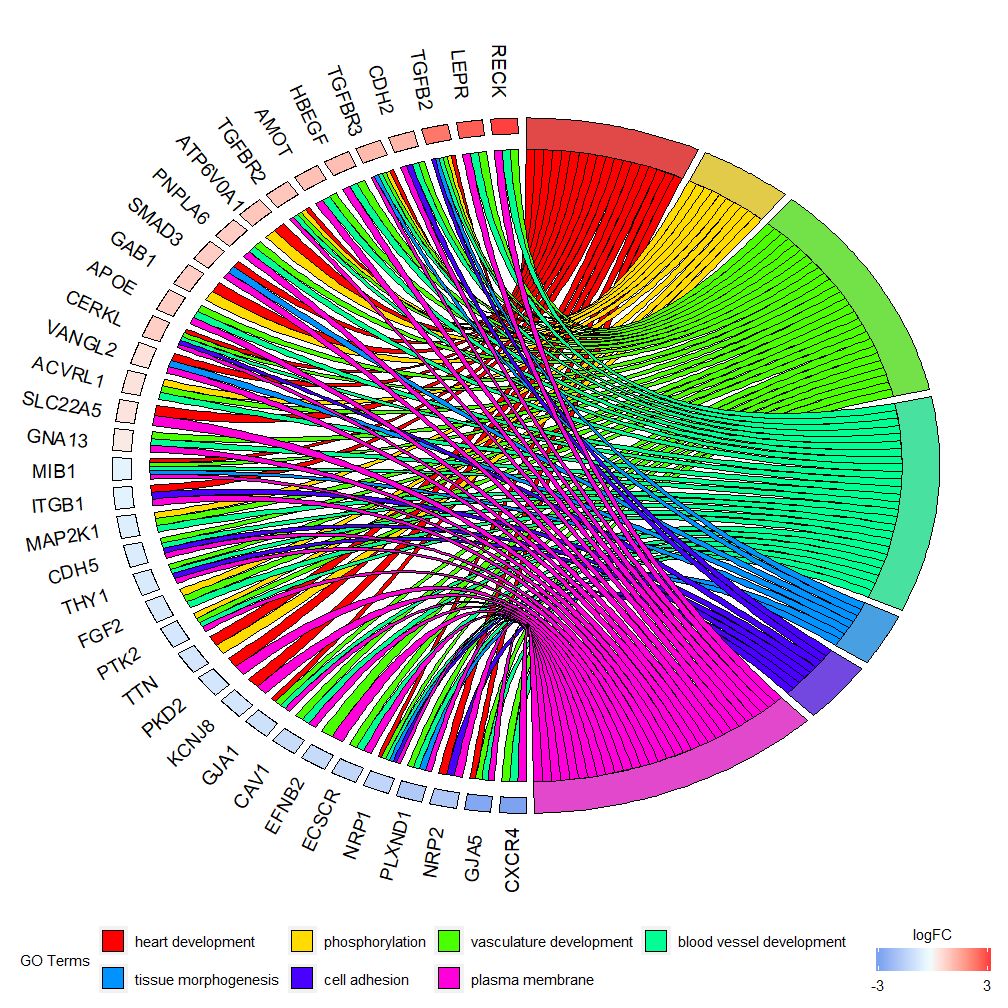
logFC 값에 따라 설정 가능gene.order=‘logFC’ , 유전자는 logFC 값에 따라 정렬됩니다. 때로는 이미지가 다소 혼잡해질 수 있으며 이는 표시되는 유전자 또는 경로의 수를 줄이기 위해 제한 매개변수를 사용하여 자동화할 수 있습니다. Limit은 두 개의 컷오프 값을 갖는 벡터입니다(기본값은 c(0,0)). 첫 번째 값은 유전자가 할당되어야 하는 최소 경로 수를 지정합니다. 두 번째 값은 경로에 할당된 유전자의 수를 결정합니다.
- # 仅显示分配给至少三个通路的基因
- GOChord(chord, limit = c(3, 0), gene.order = 'logFC')
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
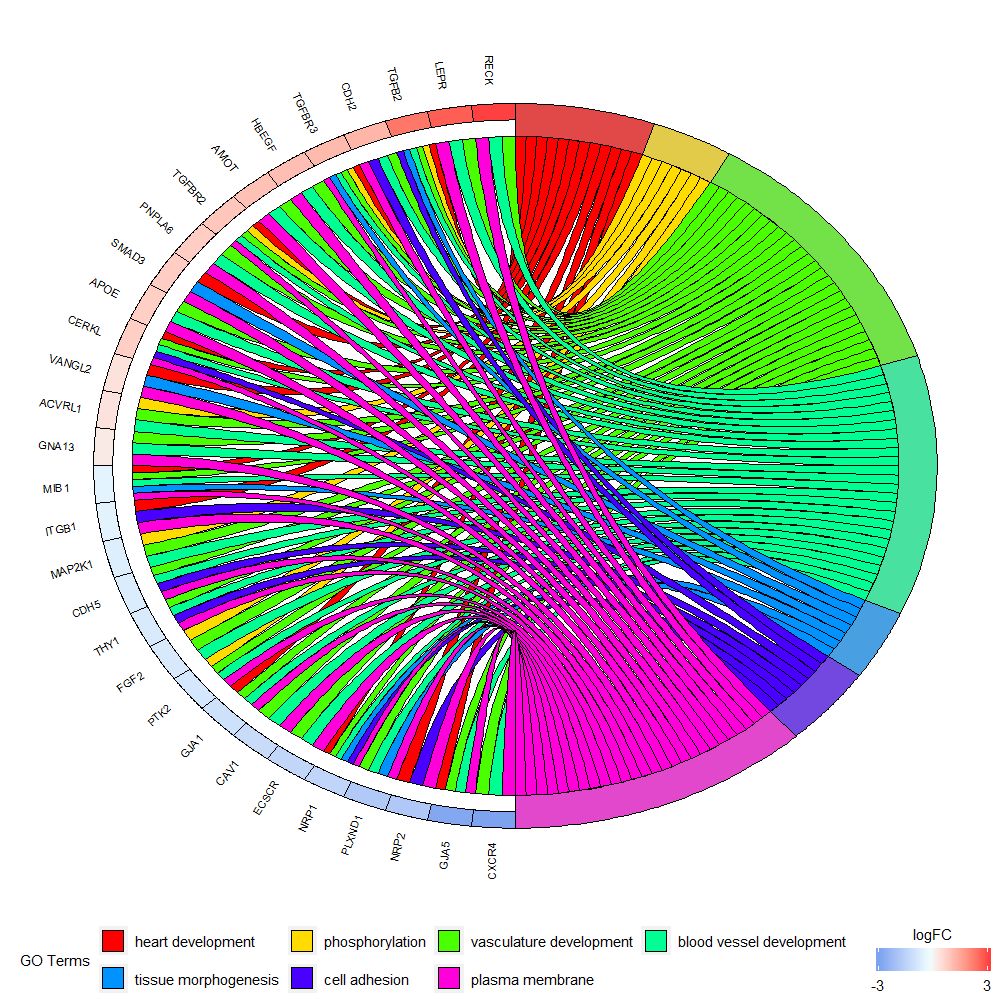
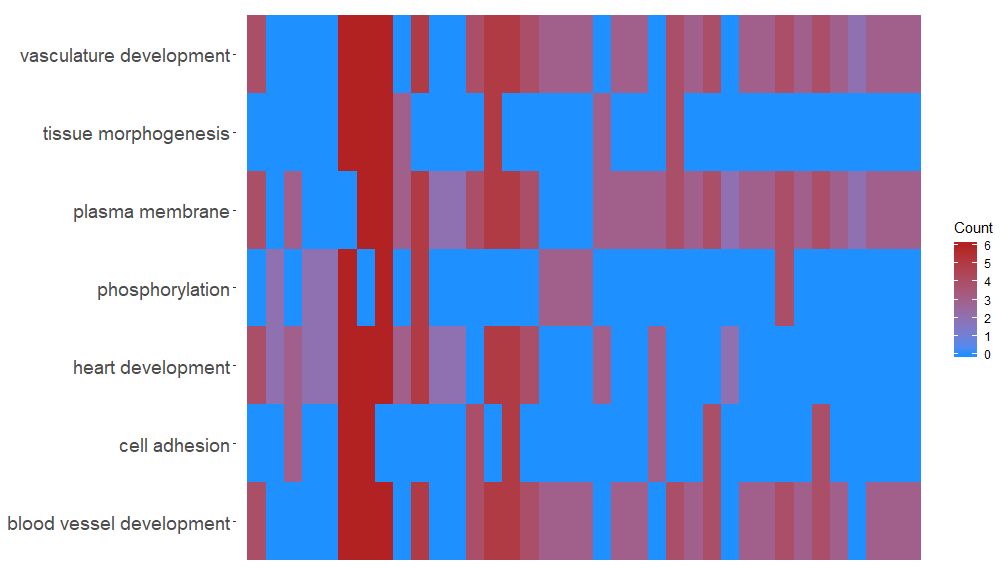
GOHeat 기능은 GOChord와 유사한 히트맵을 사용하여 유전자와 경로 사이의 관계를 표시할 수 있습니다. 생물학적 과정은 가로로 표시되고 유전자는 세로로 표시됩니다. 각 열은 작은 직사각형으로 나누어지며 색상은 일반적으로 logFC 값에 따라 달라집니다. 또한 유사한 기능적 경로가 풍부한 유전자가 클러스터링되었습니다. nlfc 매개변수에 따라 히트맵 색상 선택에는 두 가지 모드가 있습니다. nlfc = 0인 경우 색상은 각 유전자에 대한 강화된 경로의 수입니다. 자세한 내용은 예를 참조하세요.
- # First, we use the chord object without logFC column to create the heatmap
- GOHeat(chord[,-8], nlfc = 0)
GOHeat(chord[,-8])nlfc = 1인 경우 색상은 유전자의 logFC에 해당합니다.
GOHeat(chord, nlfc = 1, fill.col = c('red', 'yellow', 'green'))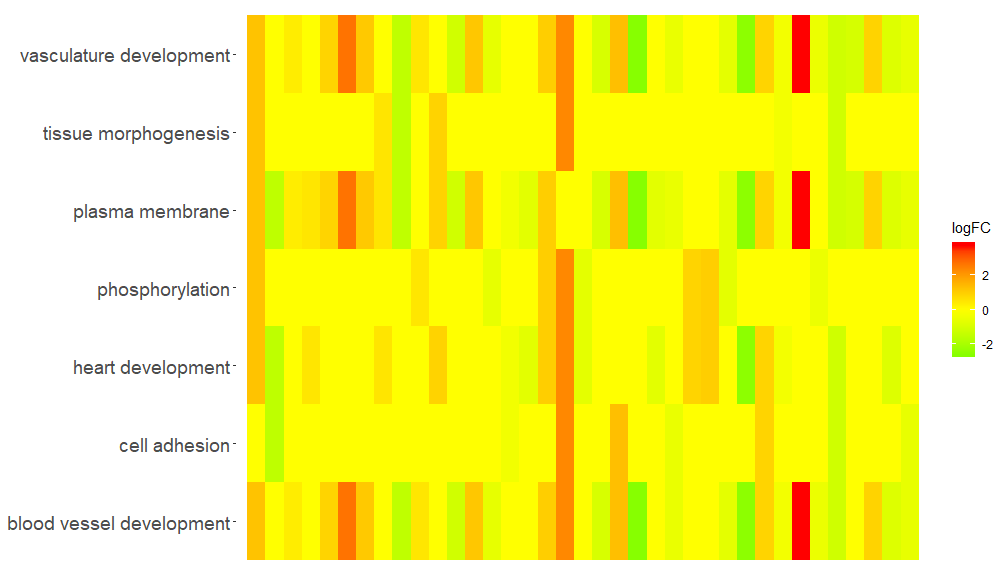
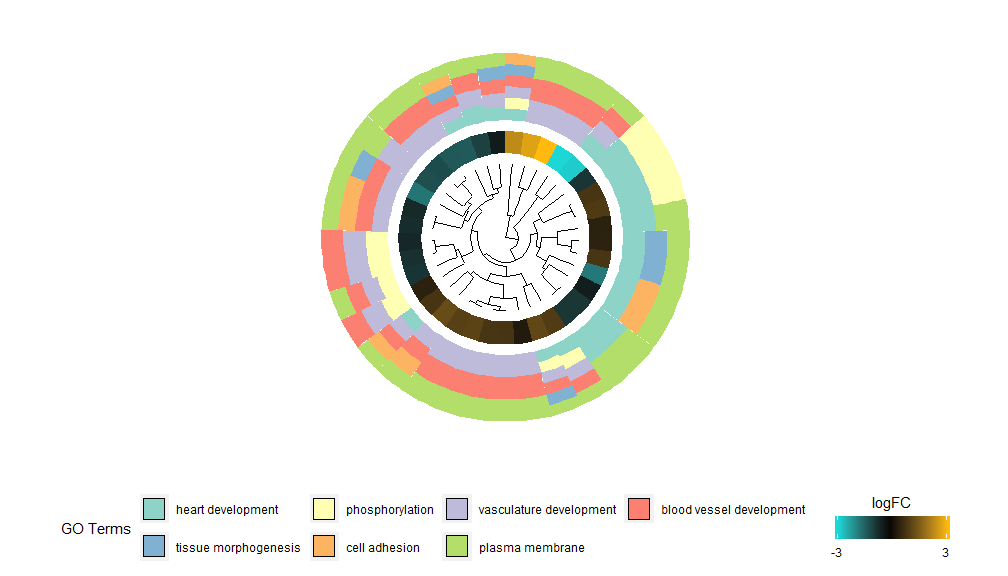
GOCluster 기능의 기본 아이디어는 가능한 한 많은 정보를 표시하는 것입니다. 예는 다음과 같습니다.
- GOCluster(circ, EC$process, clust.by = 'logFC', term.width = 2)
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
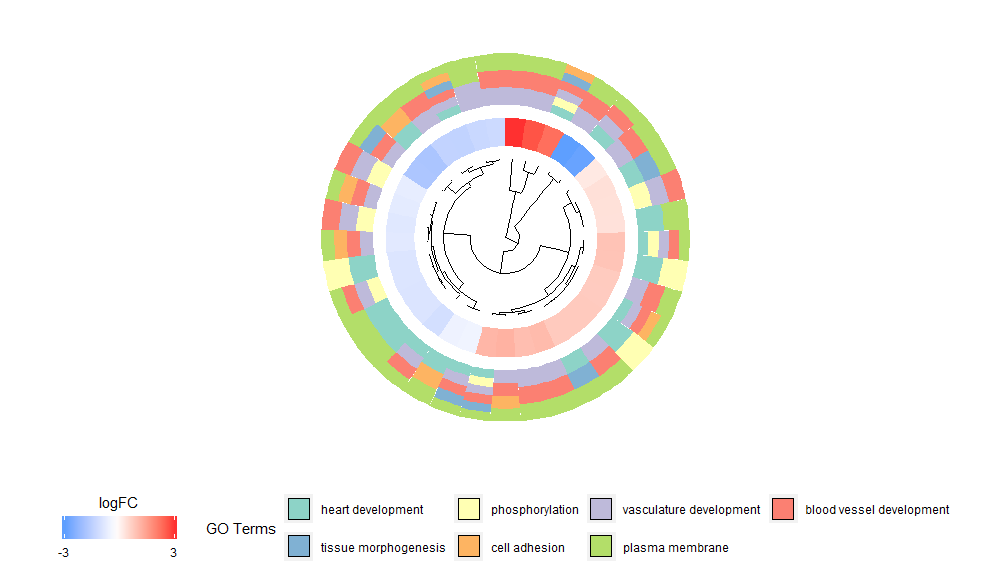
계층적 클러스터링은 발현 패턴에 따라 유전자를 편견 없이 그룹화하여 클러스터가 공동 조절되거나 기능적으로 관련된 유전자의 여러 그룹을 포함할 수 있도록 하는 유전자 발현에 대한 인기 있는 비지도 클러스터링 분석 방법입니다. GOCluster는hclust 방법은 유전자 발현 프로파일의 계층적 클러스터링을 수행합니다. 거리 측정법이나 클러스터링 알고리즘을 변경하려면 각각 metric 및 clust 매개변수를 사용하세요. 결과 덴드로그램은 ggdendro의 도움으로 변환되고 ggplot2로 시각화될 수 있습니다. 효과적일 뿐만 아니라 시각적으로도 매력적이므로 원형 레이아웃을 선택하세요. 덴드로그램 옆의 첫 번째 원은 실제로 클러스터링 트리의 잎인 유전자의 logFC를 나타냅니다. 여러 대비에 관심이 있는 경우 nlfc 매개변수를 수정할 수 있습니다. 기본적으로 이 매개변수는 "1"로 설정되어 있으므로 하나의 링만 그려집니다. logFC 값은 사용자 정의 가능한 색상 척도(lfc.col)를 사용하여 색상으로 구분됩니다. 다음 원은 유전자에 할당된 경로를 나타냅니다. 보기 좋게 보기 위해 채널 수를 줄였으며, term.col 파라미터를 이용하여 채널의 색상을 변경할 수 있습니다.아직 사용 가능?GOCluster 매개변수를 변경하는 방법을 알아보세요. 이 함수의 가장 중요한 매개변수는 Cluster.by이며, 이는 유전자 발현 패턴(위에 표시된 'logFC') 또는 기능 범주('terms')별로 클러스터링하도록 지정할 수 있습니다.
- GOCluster(circ, EC$process, clust.by = 'term', lfc.col = c('darkgoldenrod1', 'black', 'cyan1'))
-
- ## Warning: Using size for a discrete variable is not advised.
-
- ## Warning: Removed 7 rows containing missing values (geom_point).
벤 다이어그램은 차별적으로 발현되는 다양한 유전자 목록 간의 관계를 검색하거나 기능 분석에서 여러 경로 유전자의 교차점을 탐색하는 데 사용할 수 있습니다. 벤 다이어그램은 중복되는 유전자의 수뿐만 아니라 유전자의 발현 패턴(보통 상향 조절, 종종 하향 조절 또는 역조절)에 대한 정보도 표시합니다. 현재 최대 3개의 데이터 세트가 입력으로 사용됩니다. 입력 데이터에는 최소한 두 개의 열이 포함되어 있습니다. 하나는 유전자 이름용이고 다른 하나는 logFC 값용입니다.
- l1 <- subset(circ, term == 'heart development', c(genes,logFC))
- l2 <- subset(circ, term == 'plasma membrane', c(genes,logFC))
- l3 <- subset(circ, term == 'tissue morphogenesis', c(genes,logFC))
- GOVenn(l1,l2,l3, label = c('heart development', 'plasma membrane', 'tissue morphogenesis'))
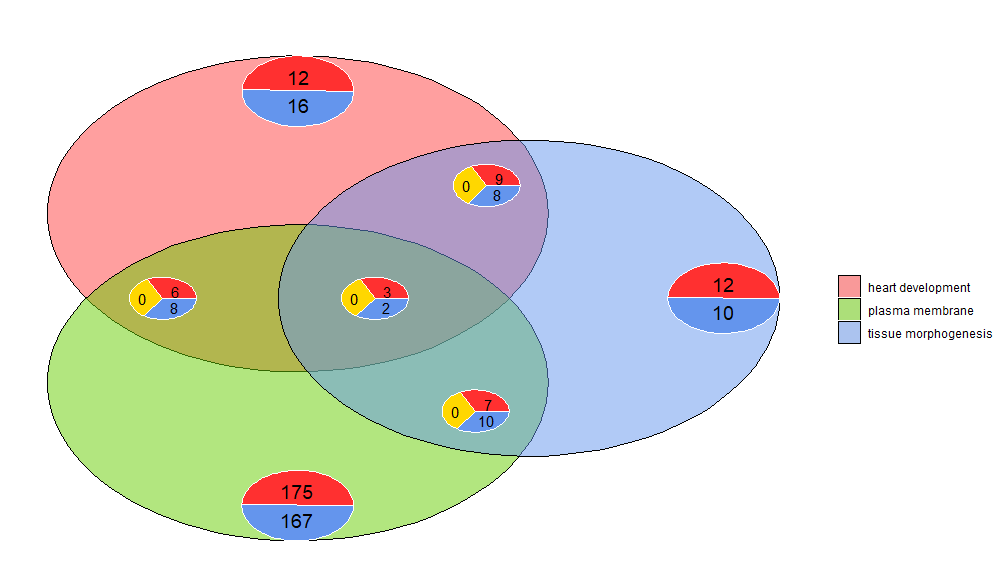
예를 들어, 심장 발달과 조직 형태형성에는 22개의 유전자가 있으며, 12개는 상향 조절되고 10개는 하향 조절됩니다. 주목해야 할 중요한 점은 원형 차트가 중복된 정보를 표시하지 않는다는 것입니다. 따라서 세 개의 데이터 세트를 비교하면 모든 데이터 세트에 공통된 유전자(가운데 파이 차트)가 다른 파이 차트에 포함되지 않습니다. 이 도구는 Shinyapp https://wwalter.shinyapps.io/Venn/에서 사용할 수 있습니다. 웹 도구는 더욱 대화형이며 원은 데이터 세트의 유전자 수에 비례하는 영역을 가지며 슬라이더를 사용하여 작은 원형 차트가 있으며 GOVenn에는 플롯 레이아웃을 변경하고 이미지 및 유전자 목록을 다운로드할 수 있는 모든 옵션이 있습니다.
소프트웨어 홈페이지: https://wencke.github.io/

그는 30년 이상 기술 연구에 전념해 왔으며, java, linux, javascript, php, css 등 다양한 언어에 능숙하며, 오픈 소스 분야에 많은 공헌을 했습니다. 나중에 참조할 수 있도록 기술 개발의 일부 문제를 공유하는 개발자 문서 스테이션입니다.

우편메소피아@프로톤메일.com